Professor
IISER Thiruvananthapuram, School of Chemistry



Our research group focuses on various aspects of Physical Organic Chemistry and Biophysical Chemistry to understand the effect of light on organic small molecules and biomolecules. Synthetic efforts to modulate the rate of charge recombination that is monitored using ultrafast spectroscopic techniques is one of the primary interests in our group at IISER Thiruvananthapuram. We also look at the strength of weak interactions in novel crystals using theoretical models such as quantum theory of atoms in molecules and exciton migration using cluster model. A combined effort to synthesise novel and diverse architectures, investigate/regulate the ultrafast processes and understand the theoretical reasons behind the events makes our attempt unique.
Mahesh Hariharan is the Professor, School of Chemistry at Indian Institute of Science Education and Research Thiruvananthapuram. He is a Fellow of the Royal Society of Chemistry and an associate editor for (i) Photochemical and Photobiological Sciences and (ii) RSC Advances. His research efforts primarily focus on understanding the interaction of light with biomolecules and crystalline organic materials.
IISER Thiruvananthapuram, School of Chemistry
University of Wuerzburg, Germany
IISER Thiruvananthapuram, School of Chemistry
Montana State University, Montana, USA
IISER Thiruvananthapuram, School of Chemistry
Northwestern University, Illinois, USA
Mentor:Prof. Frederick D. Lewis
Ph.D. in Chemistry
National Institute for Interdisciplinary Science and Technology, Trivandrum, Kerala
Title
Design of Photoactivated DNA Cleaving Agents: Synthesis and Study of Photophysical and Photobiological Properties of Bifunctional Organic Ligands
Supervisor
Dr. Danaboyina Ramaiah
Master of Science
Mahatma Gandhi University, Kottayam, Kerala
Bachelor of Science
Mahatma Gandhi University, Kottayam, Kerala

To promote internationalization, the Chemical Society of Japan established "Lectureship Award" in 2007. In 2017, 25 outstanding young chemists were selected from 9 divisions as the "2017 Lectureship Award winners" among large number of applicants. Mahesh received the award under the Photochemistry division.


Prof. Mahesh Hariharan bags Kerala State Young Scientist Award 2013 instituted by the Kerala State Council for Science, Technology and Environment (KSCSTE), in recognition of his outstanding contributions. Mahesh Hariharan had made significant contributions in several areas of photoscience that includes the fundamental understanding of effect of ultraviolet light on DNA. The award comprises a grant of Rs. 50,000, a start-up research grant (upto 50 lakhs) and travel support for presenting the research work at a conference outside the country.
We design/synthesise small molecule based donor-acceptor systems to enhance the lifetime of photoinduced charge separated states in organic molecules. Preliminary efforts led us to an understanding that organised donor or acceptor in the presence of respective counterpart (acceptor/donor) improves the survival time of charged intermediates. Attempts are in progress to create better architectures/strategies to improve the efficiency of photoinduced charge separation.
Dihydrogen and other weak interactions are utilised to tune the opto-electronic properties in crystals. We explore the rate of exciton diffusion in organic crystals of diverse packing. In summary, our group aims at understanding ultrafast processes in organic structures having diverse nearest neighbour interaction. We utilize a combination of weak interactions such C–H•••H–C, C–H•••O, C-H•••pi and pi-pi to create various interplanar angles between the adjacent arenes. Using spectroscopic techniques, we probe ultrafast processes such as energy and exciton migration across these architectures.


Achieving faster charge separation in organic systems capable of mimicking the electron transfer events in natural photosynthesis has been an exciting research topic for several decades. Herein, we demonstrate the orientation-dependent acceleration of symmetry-breaking charge separation (SB-CS) in an angular (A-PDI2) versus linear (L-PDI2) perylenediimide dimer. Femtosecond transient absorption measurements reveal ultrafast SB-CS in A-PDI2 (τCS = 6.3 ps) with charge separation ∼20 times faster than in L-PDI2 (τCS = 127.9 ps). Nanosecond transient absorption measurements establish the negligible population of triplet excited-states in L-PDI2 (ϕT < 1%), whereas a significant triplet excited-state population (ϕT = 35.9%) is quantified in A-PDI2. The theoretically computed Coulombic coupling strength in A-PDI2 (|JCoul| = 14.9 cm–1) and L-PDI2 (|JCoul| = 438.4 cm–1) is rationalized as the crucial factor modulating the SB-CS rates. The current investigation could be beneficial for designing light harvesting materials capable of faster charge separation for efficient optoelectronic devices.

Herein, we present direct evidence of intersystem crossing (ISC) in a core-twisted naphthalenemonoimide-fused perylenediimide (NP) at the single-molecule level. The ISC process was characterized by frequent quantum jumps (short off states) in the single-molecule fluorescence measurements. The singlet to triplet quantum jumps in NP was corroborated by ultrafast transient absorption measurements. NP exhibits intersystem crossing with a rate constant kISC = 6.5 × 107 s–1 and a triplet quantum yield ϕT = 25.8 ± 2.4%. Our theoretical investigations unveil the potential crossing and state mixing between first singlet excited state S1 and third triplet excited state T3, thus providing insight into the ISC mechanism during the relaxation of NP from the Franck–Condon region to its optimal geometry in the S1 state. The current investigation provides critical insights into the twist-induced ISC process in a polyaromatic hydrocarbon at both the single-molecule and ensemble levels.
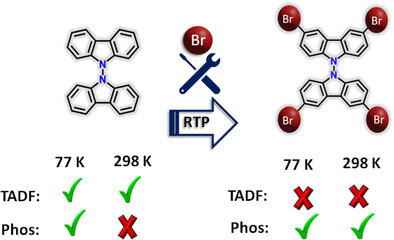
Room-temperature phosphorescence (RTP) in metal-free organic materials offers immense potential for advanced optoelectronic applications. However, the rational design and fine-tuning of RTP remain challenging due to the complex correlation between molecular structure and photophysical processes. Herein, we explored the impact of intersystem crossing (ISC), spin-orbit coupling (SOC), and halogen interactions to promote RTP in crystalline brominated carbazole dimer (BrCz-D). In contrast, the unsubstituted analogue, carbazole dimer (Cz-D) exhibits thermally activated delayed fluorescence (TADF) under ambient conditions. Femtosecond transient absorption (fsTA) spectroscopy measurements confirmed the population of triplet manifolds in both dimers. Bromine substitution significantly enhances spin-orbit coupling (VSOC = 14.94 cm⁻¹), enabling efficient ISC and robust RTP in BrCz-D. Enhanced RTP in crystalline BrCz-D is attributed to unique halogen interactions, including Br···Br, C···Br, and H···Br, within the crystal lattice. Such halogen interactions are negligible in the solution state, accounting for the lack of RTP under ambient conditions. The present work highlights the critical role of SOC and halogen bonding in achieving efficient RTP for designing high-performance organic phosphorescent materials through crystallochemistry.

Photoluminescence (PL) is a property of great interest in science and technology. Here we review recent approaches for the control of fluorescence and phosphorescence by tailored supramolecular environments. Taking recent examples from solid-state materials as applied in organic light-emitting diodes (OLEDs), supramolecular complexes, cyclophanes, foldamers, and protein–dye complexes, as well as DNA and RNA aptamers, we showcase how specific ‘smart’ matrices can modulate PL properties including brightness, quantum yield, wavelength, and lifetime. From the examples collected in this review it becomes evident that, by means of an advanced understanding of specific interactions of chromophores with the surrounding matrix, relevant processes in the excited state can be controlled in a desired way, leading to improved light-emitting materials.
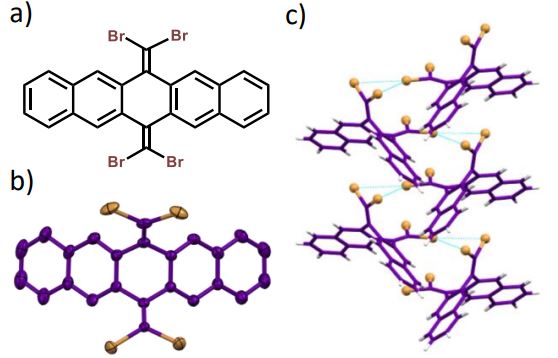
Herein, we report room-temperature phosphorescence in a brominated dihydropentacene derivative having bifurcating bromine synthon in the crystalline state. Delayed emission experiments and insights from theoretical investigations have corroborated the feasibility of long-lived triplet excitons, offering promising implications for synthon-stabilized, metal-free, organic phosphors.
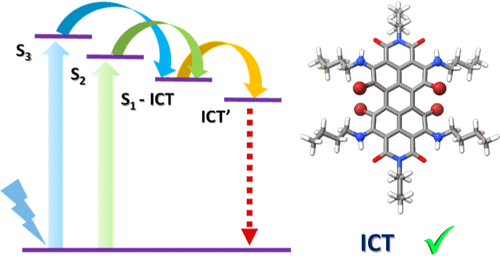
Photoinduced charge transfer (CT) states play a pivotal role in increasing the power conversion efficiency of molecular systems used in artificial photosynthesis, photocatalysis, and optronic devices. The absence of intrinsic CT states is one of the main reasons for the poor photoconversion efficiencies of organic chromophores like perylenediimide (PDI). Herein, we explore the excited state dynamics of a persubstituted PDI (AP) with amino groups at the ortho positions and bromine atoms at the bay positions. Due to the influence of bromine atoms and amino groups on the PDI core, nonradiative pathways are accessed on photoexcitation in AP. Femtosecond and nanosecond transient absorption measurements in weakly polar and polar solvents showed the relaxation of the higher singlet excited state in picoseconds time scale, paving the way to an intramolecular charge transfer (ICT) state having a lifetime in the nanoseconds time scale. As the dielectric medium changed from the weakly polar solvent (toluene, ε = 2.38) to a polar solvent (ethyl acetate, ε = 6.02), the lifetime of the solvent stabilized CT state decreased from τ = 69.1 ± 1.7 ns to τ = 47.1 ± 0.5 ns, which confirms the solvent dependency of the ICT state. Theoretical investigations employing surface hopping dynamics suggest that the rate of internal conversion (kIC = 1.13 × 1011 s–1) competes with the intersystem crossing (kISC = 0.85 × 1011 s–1) in AP. Amination on the ortho position induces the CT characteristics to the core of the PDI, as evident from hole–electron surface analysis of the S1 state. Presented results on persubstituted PDI with long-lived relaxed CT states may improve the designing strategies of organic optoelectronic devices.
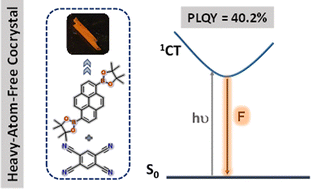
Harvesting luminescence from charge-transfer cocrystals is an efficient strategy for developing solid-state light-emitting materials without requiring the multistep organic synthesis. Herein, we report comprehensive single-crystal, computational, and spectroscopic investigations of a heavy-atom-free charge-transfer cocrystal exhibiting a 40.2% photoluminescence quantum yield.
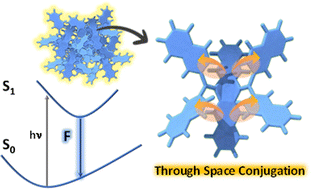
Through-space conjugation in organic chromophores offers significant potential for developing highly efficient luminescent materials. Herein, we investigate the luminescencent properties of crystalline tetra-naphthalene connected dihydropentacene isomers, 1-NP and 2-NP, using both experimental and theoretical approaches, establishing the presence of through-space conjugation mediated luminescence enhancement.
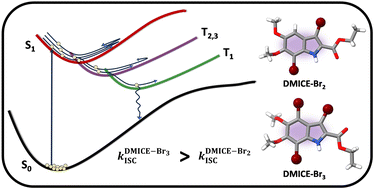
Achieving intersystem crossing (ISC) through structural tuning in biological systems is an evolving area for therapeutic and materials research. Eumelanin, a natural pigment, offers huge potential for bio-inspired material design, yet remains underexplored in this regard. Herein, we report the ultrafast intersystem crossing in di-brominated (DMICE-Br2) and tri-brominated (DMICE-Br3) eumelanin model monomers through transient absorption spectroscopy and surface hopping dynamics. Femtosecond and nanosecond transient absorption experiments suggest triplet excited state populations in DMICE-Br2 and DMICE-Br3 with triplet quantum yields and rates of ISC as Image ID:d4qo01832j-t1.gif, Image ID:d4qo01832j-t2.gif and Image ID:d4qo01832j-t3.gif, Image ID:d4qo01832j-t4.gif respectively. Theoretical insights into ISC were obtained with nonadiabatic dynamics simulations using the surface hopping including arbitrary couplings method coupled to potential energy surfaces, modelled by linear vibronic coupling (SHARC/LVC). The results show that for both DMICE-Br2 and DMICE-Br3, the initial S1 population decays to the T2 and T3 states in the picosecond timescale to further undergo internal conversion to T1 within sub-ns for DMICE-Br2 and sub-ps for DMICE-Br3. The simulated Image ID:d4qo01832j-t5.gif and Image ID:d4qo01832j-t6.gif corroborate to the assignment of the ultrafast triplet excited state population observed in the experiments. The increased triplet yields and ISC rates in DMICE-Br2 and DMICE-Br3 are attributed to the enhanced heavy atom effect from additional bromine atoms. This work presents the experimental and computational evidence for ultrafast ISC in multi-brominated eumelanin monomers, with promising implications for eumelanin-inspired material design and photodynamic applications.
Classically, aromaticity portrays the unique stability and peculiar reactivities of cyclic planar conjugated systems with (4n+2) π electrons. Understanding the electronic environments in new chemical frameworks through experimental and theoretical validation is central to this ever-expanding theme in chemical science. Such investigations in curved π-surfaces have special significance as they can unravel the variations when the planarity requirement is slightly lifted. In this report, we discuss the synthesis, spectroscopic and theoretical studies involving a new group of cyclazine analogs having a charged aza[10]annulene periphery, centrally locked through a sp3 carbon. Magnetic anisotropic effects arising from electron delocalization through its curved π-surface were mapped through a specific set of chemical groups introduced through this sp3 carbon. The nucleus-independent chemical shift calculations revealed negative chemical shift values, indicating the aromatic nature of the aza[10] annulene rim. This is corroborated by a clockwise diatropic ring current, evident from anisotropy-induced current density analysis. Variations in the chemical shift of NMR signals in these systems were also computationally examined through isotropic chemical shielding surface analysis.
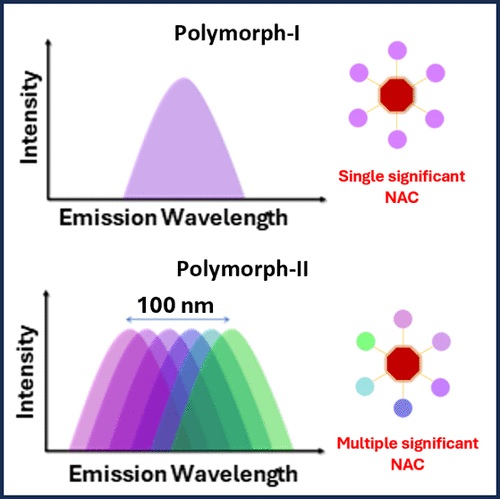
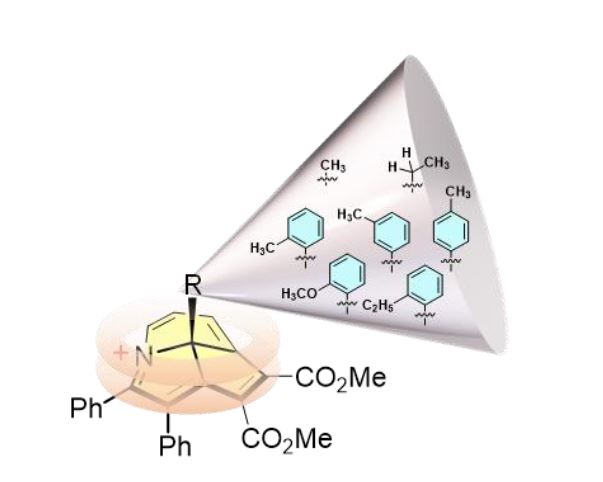
Achieving wide-range tunable emission colors in the crystalline state of single fluorophores is a challenging task. Here, we report the vibronic coupling in the crystalline state of a hitherto unexplored class of nonaromatic tetrabrominated dibenzocyclooctatetraene in two distinct polymorphic forms.The thermodynamically stable polymorph (P1) revealed a single-emission maximum upon excitation at different wavelengths in the crystalline state adhering to Kasha’s rule and lacks luminescence from triplet states. In contrast, the kinetically stable form of polymorph (P2) exhibited excitation wavelength–dependent emission with a range of Δ𝜆maxemi = 100 nm and room temperature phosphorescence. Prominent nonadiabatic couplings between the bright state and several lower singlet states in the dimeric dibenzocyclooctatetraene polymorph elucidate the excitation wavelength–dependent emission. The variable fluorescence behavior highlights the possibility of cyclooctatetraene derivatives as a novel class of multicolor luminescent materials for future optoelectronic applications.
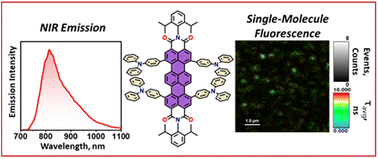
Environment-sensitive fluorescent agents with near-infrared (NIR) emission are in great demand owing to their applications in biomedical and quantum technologies. We report a novel NIR absorbing (λAbsmax = 734 nm) and emitting (λFlmax = 814 nm) terrylenediimide (TDI) based donor–acceptor chromophore (TDI-TPA4), exhibiting polarity-sensitive single-photon emission. By virtue of the charge transfer (CT) character, ensemble level measurements revealed solvatochromism and NIR emission (ϕFl = 26.2%), overcoming the energy gap law. The CT nature of the excited states is further validated by state-of-the-art fragment-based excited state theoretical analysis. To mimic the polarity conditions at the single-molecule level, TDI-TPA4 was immobilized in polystyrene (PS; low polar) and poly(vinyl alcohol) (PVA; high polar) matrices, which enables tuning of the energy levels of the locally excited state and charge-separated (CS) state. Minimal blinking and prolonged survival time of the TDI-TPA4 molecule in the PS matrix, in contrast to the PVA matrix, possibly confirms the implication of the energy gap law and polarity sensitivity of TDI-TPA4. The existence of the CT state in nonpolar and CS state in polar solvents was confirmed by transient absorption measurements in the femtosecond regime. The current work sheds light on the design principle for NIR single-photon emitting organic chromophores for deep tissue imaging and probing the nanoscale heterogeneity.
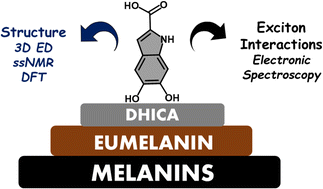
Eumelanin, a versatile biomaterial found throughout the animal kingdom, performs essential functions like photoprotection and radical scavenging. The diverse properties of eumelanin are attributed to its elusive and heterogenous structure with DHI (5,6-dihydroxyindole) and DHICA (5,6-dihydroxyindole-2-carboxylic acid) precursors as the main constituents. Despite DHICA being recognized as the key eumelanin precursor, its crystal structure and functional role in the assembled state remain unknown. Herein, we employ a synthesis-driven, bottom-up approach to elucidate the structure and assembly-specifics of DHICA, a critical building block of eumelanin. We introduce an interdisciplinary methodology to analyse the nanocrystalline assembly of DHICA, employing three-dimensional electron diffraction (3D ED), solid-state NMR and density functional theory (DFT), while correlating the structural aspects with the electronic spectroscopic features. The results underscore charge-transfer exciton delocalization as the predominant energy transfer mechanism within the π–π stacked and hydrogen-bonded crystal network of DHICA. Additionally, extending the investigation to the 13C-labelled DHICA-based polymer improves our understanding of the chemical heterogeneity across the eumelanin pigment, providing crucial insights into the structure of eumelanin.
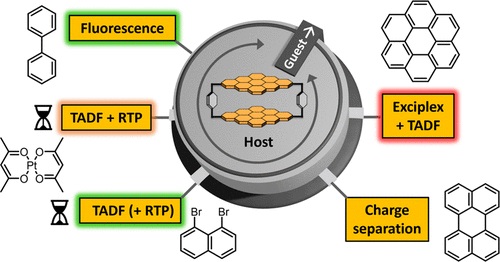
The properties and functions of chromophores utilized by nature are strongly affected by the environment formed by the protein structure in the cells surrounding them. This concept is transferred here to host–guest complexes with the encapsulated guests acting as an environmental stimulus. A new cyclophane host based on coronene bisimide is presented that can encapsulate a wide variety of planar guest molecules with binding constants up to (4.29 ± 0.32) × 10^10 M^(–1) in chloroform. Depending on the properties of the chosen guest, the excited state deactivation of the coronene bisimide chromophore can be tuned by the formation of host–guest complexes toward fluorescence, exciplex formation, charge separation, room-temperature phosphorescence (RTP), or thermally activated delayed fluorescence (TADF). The photophysical processes were investigated by UV/vis absorption, emission, and femto- and nanosecond transient absorption spectroscopy. To enhance the TADF, two different strategies were used by employing suitable guests: the reduction of the singlet–triplet gap by exciplex formation and the external heavy atom effect. Altogether, by using supramolecular host–guest complexation, a versatile multimodal chromophore system is achieved with the coronene bisimide cyclophane.
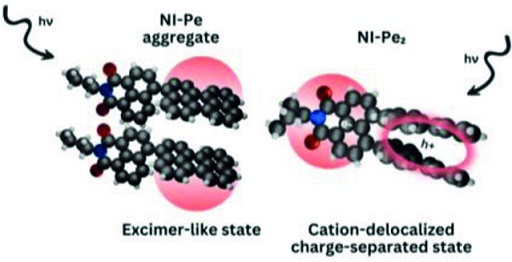
The investigation of impact of through-space/through-bond electronic interaction among chromophores on photoexcited-state properties has immense potential owing to the distinct emergent photophysical pathways. Herein, the photoexcited-state dynamics of homo-sorted π-stacked aggregates of a naphthalenemonoimide and perylene-based acceptor-donor (NI-Pe) system and a fork-shaped acceptor-bisdonor (NI-Pe2) system possessing integrally stacked peri-substituted donors was examined. Femtosecond transient absorption (fsTA) spectra of NI-Pe monomer recorded in chloroform displayed spectroscopic signatures of the singlet state of Pe; 1Pe*, the charge-separated state; NI−⋅-Pe+⋅, and the triplet state of Pe; 3Pe*. The examination of ultrafast excited-state processes of NI-Pe aggregate in chloroform revealed faster charge recombination (mathematical equation =1.75 ns) than the corresponding monomer (mathematical equation =2.46 ns) which was followed by observation of a broad structureless band attributed to an excimer-like state. The fork-shaped NI-Pe2 displayed characteristic spectroscopic features of the NI radical anion (λmax~450 nm) and perylene dimer radical cation (λmax~520 nm) upon photoexcitation in non-polar toluene solvent in the nanosecond transient absorption (nsTA) spectroscopy. The investigation highlights the significance of intrinsic close-stacked arrangement of donors in ensuring a long-lived photoinduced charge-separated state (mathematical equation =1.35 μs) in non-polar solvents via delocalization of radical cation between the donors.
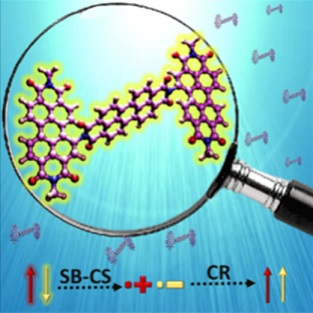
Herein, we demonstrate triplet excited-state population in a conformationally rigid perylenediimide trimer (PDI-T) via intramolecular symmetry-breaking charge separation (SB-CS) at the single-molecule level. The single-molecule fluorescence intensity trajectories of PDI-T in nonpolar polystyrene matrix (ε = 2.60) exhibit prolonged fluorescence with infrequent dark states, representing the triplet and/or the charge transfer states. In contrast, in a poly(vinyl alcohol) matrix (ε = 7.80), erratic blinking dynamics resulting in low photon counts were observed, corroborating the feasibility of charge separation in a polar environment. In agreement with the single-molecule measurements, transient absorption spectroscopy of PDI-T reveals ultrafast SB-CS (τCS < 5 ps) in polar tetrahydrofuran (ε = 7.58) and acetone (ε = 20.70), with the population of the triplet excited-state through charge recombination. The current investigation shows the utility of rigid and weakly coupled molecular constructs in controlling triplet generation and SB-CS for potential applications in optoelectronic devices.
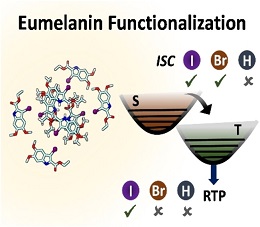
We report the room temperature phosphorescence upon iodination on a crystalline eumelanin monomer with shielded hydroxyl moieties, ethyl 5,6-dimethoxyindole-2-carboxylate (DMICE). Ultrafast intersystem crossing (ISC) is observed in the iodinated (IDMICE) as well as brominated (BDMICE) analogues of the eumelanin monomer derivative in solution. The triplet quantum yields (φT) and intersystem crossing rates (kISC) of the halogenated eumelanin derivatives are mathematical equation =25.4±1.1 %; mathematical equation =1.95×109 s−1 and mathematical equation =59.1±1.6 %; mathematical equation 1.36×1010 s−1, as monitored using transient absorption spectroscopy. Theoretical calculations based on nuclear ensemble method reveal that computed kISC and spin-orbit coupling matrix elements for eumelanin derivatives are larger for IDMICE relative to BDMICE. The halogen and π-π interactions, with distinct excitonic coupling and higher ISC rate promote phosphorescence in IDMICE molecular crystals. Accessing triplet excited states and resultant photoluminescence through structural modification of eumelanin scaffolds paves way for exploring the versatility of eumelanin-inspired molecules as bio-functional materials..
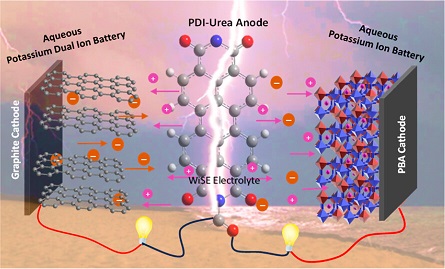
Aqueous batteries are considered as promising alternative power sources due to their eco-friendly, cost-effective, and nonflammable attributes. Employing organic-based electrode materials offers further advantages toward building greener and sustainable systems, owing to their tunability and environmental friendliness. In order to enhance the energy and power densities, superconcentrated aqueous electrolytes, such as water-in-salt electrolytes (WiSE), have renewed the interest in aqueous batteries due to their enhanced stability and much wider electrochemical stability window (>1.23 V) compared with the traditional aqueous electrolytes. Here, we present a perylene diimide-based electrode material (PDI-Urea) as an appealing anode for aqueous potassium energy storage systems and investigate their electrochemical performance in three WiSE electrolytes, namely, 30 M potassium acetate, 40 M potassium formate and 30 M potassium bis(fluorosulfonyl)imide (KFSI). To explore the potential of PDI-Urea for potassium-based electrochemical energy systems, we fabricated full cell devices such as aqueous potassium dual-ion battery (APDIB) and aqueous K-ion battery (AKIB) and studied their electrochemical properties with 30 M KFSI electrolyte. The full cell K-ion battery, using a PBA cathode, exhibited excellent electrochemical performance with good rate capability and impressive capacity retention of 91% upon 1000 cycles. Further, the reaction mechanism of the electrodes is systematically analyzed using ex-situ studies.
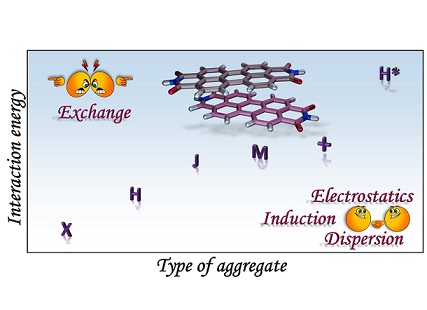
Understanding the self-assembly of conjugated organic materials at the molecular level is crucial in their potential applications as active components in electronic and optoelectronic devices. The type of aggregation significantly influences the intriguing electronic and optical characteristics differing from their constituent molecules. Perylenediimides (PDIs), electron-deficient molecules exhibiting remarkable n-type semiconducting properties, are among the most explored organic fluorescent materials due to their high fluorescence efficiency, photostability, and optoelectronic properties. PDI derivatives are reported to form well-tailored supramolecular architectures: cofacial with minor slip (H-aggregates), staggered with major slip (J-aggregates), magic angle stacking (M-aggregates), rotated (X-aggregates), rotated orthogonal ((+)-aggregates), etc. H*-aggregates are defined here as an ideal case of H-aggregate with an eclipsed configuration. Although numerous reports regarding the formation and optical properties of various PDI aggregates are known, the key driving force within the PDI units guiding the self-assembly to form distinct aggregate systems remains elusive. To unravel the molecular-level mechanisms behind the self-assembly of PDI units by probing the intermolecular interactions, symmetry-adapted perturbation theory-based energy decomposition, potential energy surface scans, and non-covalent interaction index analyses were employed on PDI dimer models. Quantum theory of atoms in molecules and frontier molecular orbital analyses were implemented on the dimer models to comprehend the effect of heteroatoms and orbital interactions in stabilising the X-aggregates over the other PDI aggregate systems. Competition between the attractive and repulsive non-covalent interactions dictates a stability order of X>H>J>M>(+)>H* for the PDI aggregate system, while in the parent perylene system, the stability order was found to be X>(+)>H>M>J>H*.
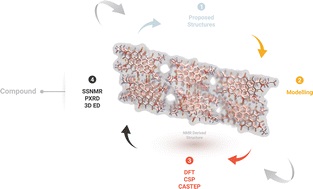
Profound knowledge of the molecular structure and supramolecular organization of organic molecules is essential to understand their structure–property relationships. Herein we demonstrate the packing arrangement of partially disordered nitro-perylenediimide (NO2-PDI), revealing that the perylenediimide units exhibit an X-shaped packing pattern. The packing of NO2-PDI is derived using a complementary approach that utilises solid-state NMR (ssNMR) and 3D electron diffraction (3D ED) techniques. Perylenediimide (PDI) molecules are captivating due to their high luminescence efficiency and optoelectronic properties, which are related to supramolecular self-assembly. Increasing the alkyl chain length on the imide substituent poses a more significant challenge in crystallizing the resulting molecule. In addition to the alkyl tails, other functional groups, like the nitro group attached as a bay substituent, can also cause disorder. Such heterogeneity could lead to diffuse scattering, which then complicates the interpretation of diffraction experiment data, where perfect periodicity is expected. As a result, there is an unmet need to develop a methodology for solving the structures of difficult-to-crystallize materials. A synergistic approach is utilised in this manuscript to understand the packing arrangement of the disordered material NO2-PDI by making use of 3D ED, ssNMR and density functional theory calculations (DFT). The combination of these experimental and theoretical approaches provides great promise in enabling the structural investigation of novel materials with customized properties across various applications, which are, due to the internal disorder, very difficult to study by diffraction techniques. By effectively addressing these challenges, our methodology opens up new avenues for material characterization, thereby driving exciting advancements in the field.
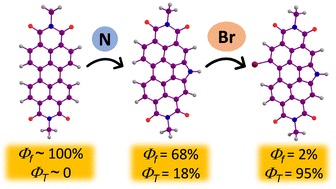
The efficient population of the triplet excited states in heavy metal-free organic chromophores has been one of the long-standing research problems to molecular photochemists. The negligible spin–orbit coupling matrix elements in the purely organic chromophores and the large singlet–triplet energy gap (ΔES–T) pose a hurdle for ultrafast intersystem crossing (ISC). Herein we report the unprecedented population of triplet manifold in a series of nitrogen-annulated perylene bisimide chromophores (NPBI and Br-NPBI). NPBI is found to have a moderate fluorescence quantum yield (Φf = 68 ± 5%), whereas Br-NPBI showcased a low fluorescence quantum yield (Φf = 2.0 ± 0.6%) in toluene. The femtosecond transient absorption measurements of Br-NPBI revealed ultrafast ISC (kISC = 1.97 × 1010 s−1) from the initially populated singlet excited state to the long-lived triplet excited states. The triplet quantum yields (ΦT = 95.2 ± 4.6% for Br-NPBI, ΦT = 18.7 ± 2.3% for NPBI) calculated from nanosecond transient absorption spectroscopy measurements showed the enhancement in triplet population upon bromine substitution. The quantum chemical calculations revealed the explicit role of nitrogen annulation in tuning the excited state energy levels to favor the ISC. The near degeneracy between the singlet and triplet excited states observed in NPBI and Br-NPBI (ΔES–T = −0.01 eV for NPBI, ΔES–T = 0.03 eV for Br-NPBI) facilitates the spin flipping in the molecules. Nitrogen annulation emerges as a design strategy to open up the ISC pathway and the rate of which can be further enhanced by the substitution of a heavier element.
Theoretical investigations on the influence of graphene fragments on the antiaromaticity of pentalene are conducted by employing multiple aromaticity descriptors based on magnetic, geometric and electronic criteria. NICS as a sole descriptor for analysing the antiaromaticity of pentalene on graphene fragments has to be carefully considered while looking through the other aromaticity indicators.
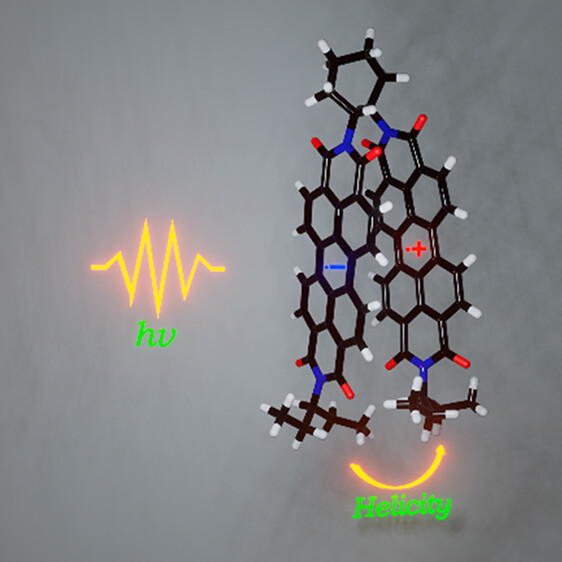
Chiral molecular assemblies exhibiting symmetry-breaking charge separation (SB-CS) are potential candidates for the development of chiral organic semiconductors. Herein, we explore the excited-state dynamics of a helically chiral perylenediimide bichromophore (Cy-PDI2) exhibiting SB-CS at the ensemble and single-molecule levels. Solvent polarity-tunable interchromophoric excitonic coupling in chiral Cy-PDI2 facilitates the interplay of SB-CS and excimer formation in the ensemble domain. Analogous to the excited-state dynamics of Cy-PDI2 at the ensemble level, single-molecule fluorescence lifetime traces of Cy-PDI2 depicted long-lived off-states characteristic of the radical ion pair-mediated dark states. The discrete electron transfer and charge separation dynamics in Cy-PDI2 at the single-molecule level are governed by the distinct influence of the local environment. The present study aims at understanding the fundamental excited-state dynamics in chiral organic bichromophores for designing efficient chiral organic semiconductors and applications toward charge transport materials.
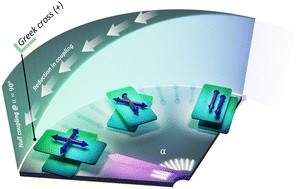
Fundamental understanding of the supramolecular assemblies of organic chromophores and the development of design strategies have seen endless ripples of interest owing to their exciting photophysical properties and optoelectronic applications. The independent discovery of dye aggregates by Jelley and Scheibe was the commencement of the remarkable advancement in the field of aggregate photophysics. Subsequent research warranted an exceptional model for defining the exciton interactions in aggregates, proposed by Davydov, Kasha and co-workers, independently, based on the long-range Coulombic coupling. Fascinatingly, the orthogonally cross-stacked molecular transition dipole arrangement was foretold by Kasha to possess null exciton interaction leading to spectroscopically uncoupled molecular assembly, which lacked an experimental signature for decades. There have been several attempts to identify and probe atypical molecular aggregates for decoding their optical behaviour. Herein, we discuss the recent efforts in experimentally verifying the unusual exciton interactions supported with quantum chemical computations, primarily focusing on the less explored null exciton splitting. Exciton engineering can be realized through synthetic modifications that can additionally offer control over the assorted non-covalent interactions for orchestrating precise supramolecular assembly, along with molecular editing. The task of attaining a minimal excitonic coupling through an orthogonally cross-stacked crystalline architecture envisaged to offer a monomer-like optical behaviour was first reported in 1,7-dibromoperylene-3,4,9,10-tetracarboxylic tetrabutylester (PTE-Br2). The attempt to stitch molecules covalently in an orthogonal fashion to possess null excitonic character culminated in a spiro-conjugated perylenediimide dimer exhibiting a monomer-like spectroscopic signature. The computational and experimental efforts to map the emergent properties of the cross-stacked architecture are also discussed here. Using the null aggregates formed by the interference effects between CT-mediated and Coulombic couplings in the molecular array is another strategy for achieving monomer-like spectroscopic properties in molecular assemblies. Moreover, identifying supramolecular assemblies with precise angle-dependent properties can have implications in functional material design, and this review can provide insights into the uncharted realm of null exciton splitting.
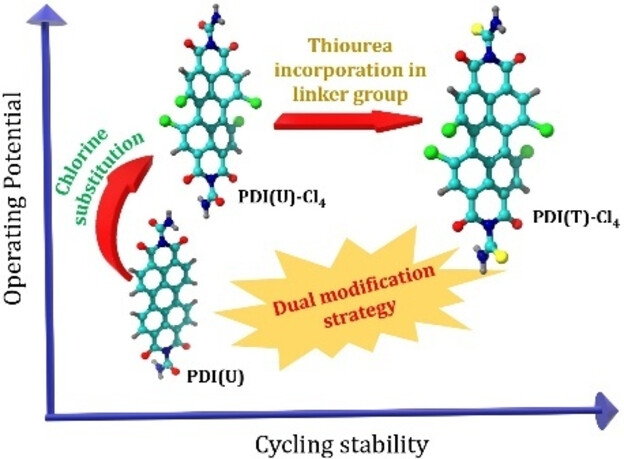
Establishing a sustainable energy solution is one of the most important issues in achieving a greener community. Given the geographically limited cobalt resources, the rising concerns about the growth of electric vehicle sectors driven by lithium-ion batteries consisting of cobalt-based cathodes have pushed the research community to probe alternate avenues. In this endeavor, employing organic electrodes can open the road to green and sustainable batteries. Among many new emerging candidates, polyimides are still being considered as cathode candidates owing to their tunability. This work envisaged tailoring the reduction potential and enhancing the cycling stability of a perylene polyimide-based organic cathode through a dual modification strategy. The influence of the twist induced by chlorine functionality at the bay position of perylene diimide and the role of the linker is systematically summarized. The correlation between the reduction potentials and the electron-withdrawing ability of the four-chlorine bay-substituent was supported by HOMO-LUMO energy levels and orbital iso-surface studies of the monomers. The accompanying thiourea linker group has significantly increased the cycling stability for 200 cycles at 1 A g−1 current density. This approach can be expanded to other organic battery chemistries for significantly enhanced electrochemical performance.

Structure-property-function relationships play a key role in governing physical properties in molecules and materials in the broader sense. Particularly in organic light-harvesting materials, the sensitivity is magnified as the vibronic and electronic energy landscapes are highly sensitive to the type and extent of quantum chemical communication between molecules. In this article, we select a particular geometric feature- the interchromophoric twist and discuss its implication on several fundamental photophysical properties in connection with our lab’s research interests. We highlight two important processes, singlet fission (SF) and symmetrybreaking charge separation (SB-CS). We narrate how the interchromophoric rotation angle can be a crucial tool in achieving an appropriate electronic coupling in each of the cases and help us utilize these processes to achieve longlived excitons (triplets and unbound electron-hole pairs) for the design and development of efficient artificial solar energy systems.
Multitudes of novel π-conjugated materials have gained tremendous attention with potential applications in next-generation electronic devices as they are chemically programmable, mechanically flexible, and lightweight. Modulating the intermolecular interactions emerging from the π-conjugated molecules govern crystal packing and plays a pivotal role in designing molecular organic semiconductors. Determining the dynamic origin of charge transport in organic crystals from a molecular perspective has often proved challenging. In this Perspective, we have compiled effective methods to regulate the charge transport characteristics of functional organic materials in their crystalline phase. The ardent efforts in designing advanced materials with desirable charge transport properties from a theoretical perspective by our group are emphasized.
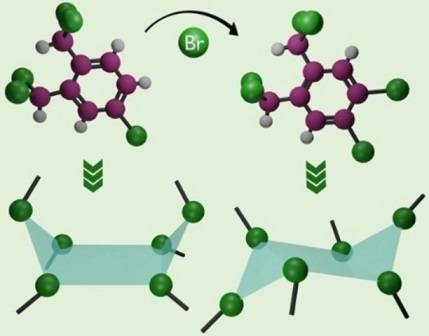
Non-covalent halogen bonding interactions are quintessential in crystal engineering for the construction of distinctive supramolecular synthons. Here, we report the first crystalline evidences of unique boat and chair shaped cyclic hexahalogen synthons in the crystal structures of α,α,α′,α′,4-pentabromo-o-xylene (PBX) and α,α,α′,α′,4,5-hexabromo-o-xylene (HBX) respectively. Nature and stability of constituent interactions in the supramolecular synthons are scrutinized with the help of quantum-chemical calculations. Pendás’ interacting quantum atoms approach confirmed the stability of Br⋅⋅⋅Br interactions leading to boat and chair shaped synthons with major contribution from exchange-correlation. Although both the molecules are achiral in nature, the packing forces guide PBX to crystallize in the chiral space group P21 with a helix-like orientation while HBX packs in a centrosymmetric P21/n space group. The extended furcations in the pentabromo derivative construct a molecular framework consisting of macrocycles realized through halogen bonding.
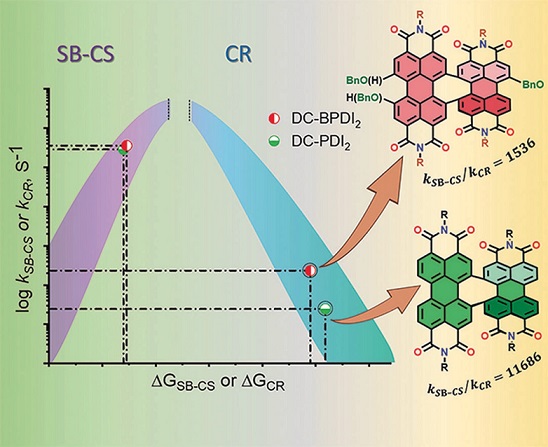
We report a long-lived charge-separated state in a chromophoric pair (DC-PDI2) that uniquely integrates the advantages of fundamental processes of photosynthetic reaction centers: i) Symmetry-breaking charge-separation (SB-CS) and ii) Marcus-inverted-region dependence. The near-orthogonal bichromophoric DC-PDI2 manifests an ultrafast evolution of the SB-CS state with a time constant of urn:x-wiley:14337851:media:anie202216482:anie202216482-math-0001 =0.35±0.02 ps and a slow charge recombination (CR) kinetics with urn:x-wiley:14337851:media:anie202216482:anie202216482-math-0002 =4.09±0.01 ns in ACN. The rate constant of CR of DC-PDI2 is 11 686 times slower than SB-CS in ACN, as the CR of the PDI radical ion-pair occurs in the deep inverted region of the Marcus parabola (urn:x-wiley:14337851:media:anie202216482:anie202216482-math-0003 >λ). In contrast, an analogous benzyloxy (BnO)-substituted DC-BPDI2 showcases a ≈10-fold accelerated CR kinetics with urn:x-wiley:14337851:media:anie202216482:anie202216482-math-0004 lowering to ≈1536 in ACN, by virtue of a decreased CR driving force. The present investigation demonstrates a control of molecular engineering to tune the energetics and kinetics of the SB-CS material, which is essential for next-generation optoelectronic devices.
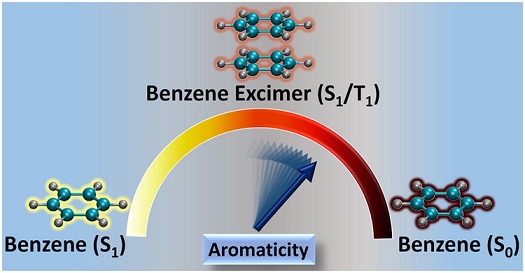
Excited state aromaticity is a stimulating area of research, widely used as a probe to describe and rationalize many photochemical phenomena. Herein, we review some of the recent findings of unprecedented aromatic stabilization in spin singlet excimer and through-space aromatic character in triplet excimers of a series of linear [n]acenes, as paramount examples of polycyclic aromatic hydrocarbons (PAHs). This review also provides insights on the aromatic stabilization profile of singlet benzene excimer formation, which can be related to antiaromaticity alleviation of the molecular (localized) S1 through exciton delocalization. The theoretical investigation of excimer stabilization using magnetic, electronic, and geometric aromatic indices manifested the presence of through-space ring current in triplet cofacial excimers. The antiaromaticity of the sandwich (D6h) spin singlet and triplet benzene excimers was also investigated by decomposing the excimer wave function as a linear combination of local exciton (LE) and charge transfer (CT) diabats and by identifying the contribution of these terms to the nucleus independent chemical shift (NICS) of the two 6-membered rings. These results provide a detailed description of the unique (anti)aromatic properties in PAH excimers, establishing strong connection between this important chemical concept and the electronic structure intricacies of excimers.
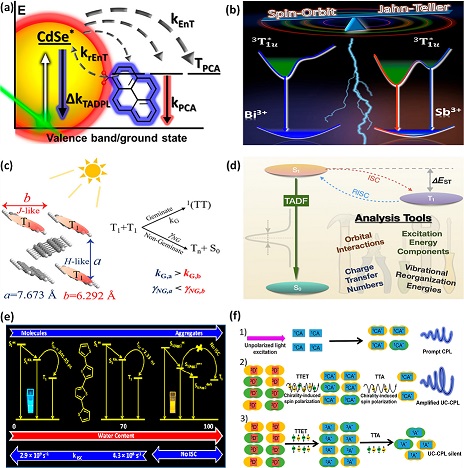
Triplet excited states find multiple potential applications in the fields of photocatalysis, photodynamic therapy, molecular logic gates, photovoltaics, and triplet–triplet annihilation up-conversion processes. The requirement of longer exciton lifetimes in the development of efficient optoelectronic devices has seen an incredible rise in the number of research articles on triplet excited states in recent years. This Virtual Issue (VI) highlights recent publications from The Journal of Physical Chemistry Letters that describe research on triplet excitons and their applications. The systems emphasized in this VI include organic host–guest materials, thermally activated delayed fluorescence (TADF) materials, room-temperature phosphorescence (RTP) materials, pure organic doped systems, organic semiconductors, quantum dots (QDs), and hybrid perovskites.
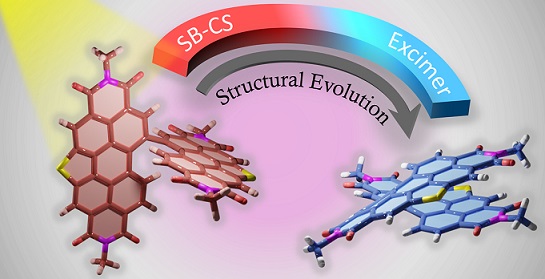
Achieving long-lived symmetry-broken charge-separated state in chromophoric assemblies is quintessential for enhanced performance of artificial photosynthetic mimics. However, the occurrence of energy trap states hinders exciton and charge transport across photovoltaic devices, diminishing power conversion efficiency. Herein, we demonstrate unprecedented excimer formation in the relaxed excited-state geometry of bichromophoric systems impeding the lifetime of symmetry-broken charge-separated states. Core-annulated perylenediimide dimers (SC-SPDI2 and SC-NPDI2) prefer near-orthogonal arrangement in the ground state and a π-stacked foldamer structure in the excited state. The prospect of an excimer-like state in the foldameric arrangement of SC-SPDI2 and SC-NPDI2 has been rationalized by fragment-based excited state analysis and temperature-dependent photoluminescence measurements. Effective electronic coupling matrix element in the Franck-Condon geometry of SC-SPDI2 and SC-NPDI2 facilitate solvation-assisted ultrafast symmetry-breaking charge-separation (SB-CS) in a high dielectric environment, in contrast to unrelaxed excimer formation (Ex*) in low dielectric environment. Subsequently, the SB-CS state dissociates into an undesired relaxed excimer state (Ex) due to configuration mixing of Frenkel exciton (FE) and charge-separated state in the foldamer structure, downgrading the efficacy of the charge-separated state. The decay rate constant of FE to SB-CS (kFE→SB–CS) in polar solvents is 8-17 fold faster than direct Ex* formation (kFE→Ex*) in non-polar solvent (kFE→SB–CS≫kFE→Ex*), characterized by femtosecond transient absorption (fsTA) spectroscopy. The present investigation establishes the impact of detrimental excimer formation on the persistence of SB-CS state in chromophoric dimers and offers the requisite of conformational rigidity as one of the potential design principles for developing advanced molecular photovoltaics.
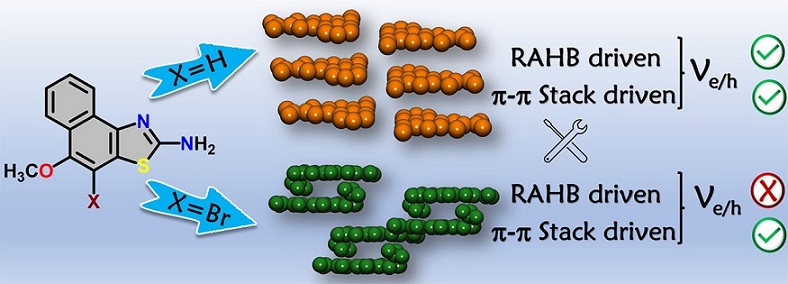
Supramolecular chemistry employs noncovalent interactions to coax π-conjugated molecules into ordered functional assemblies. Herein, we report 5-methoxynaphtho[1,2-d]thiazol-2-amine (NTN) and 4-bromo-5-methoxynaphtho[1,2-d]thiazol-2-amine (NTNB) assembled into π-stacked columns integrated by lateral resonance-assisted hydrogen bonds (RAHBs) orthogonal to the π–π stacking direction. Quantum theory of atoms in molecules (QTAIM) and interacting quantum atoms (IQA) analyses were utilized to characterize the presence and stability of intermolecular RAHBs in NTN and NTNB. In contrast to the parallel packing of NTN, bromine substitution promoted antiparallel packing in NTNB with noticeable π–π stacking and orbital overlap favoring efficient charge transfer coupling (Ve/h). Antiparallel stacking in NTNB exhibits a dipole moment minimization and aromaticity gain. The relevance of aromaticity in stabilizing π–π stacked systems is highlighted by the aromaticity gain in antiparallel stacked dimers of NTNB and can be extended to estimate the nature and strength of noncovalent interactions. Crystal packing plays a crucial role in regulating the charge transport properties, as can be observed from higher electron and hole transfer coupling along the π–π stacked and RAHB dimer, respectively, in NTN. However, in NTNB maximum electron and hole transfer coupling occurs selectively along the π–π stacked antiparallel dimer. The anisotropic mobility plots from a combination of first-principles quantum chemical calculations and the Marcus–Hush formalism confirm that both RAHB and π–π stacked dimers contribute to the mobility in NTN, but NTNB exclusively benefits from the π–π stacked dimer. Modulating noncovalent interactions for charge carrier transport can harness the innate potential of organic molecules to engineer novel optoelectronic materials.
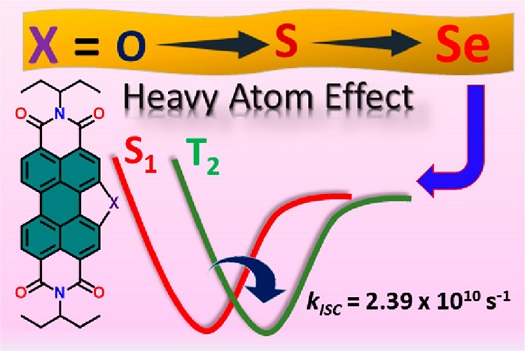
An intersystem crossing (ISC), the non-radiative transition between two electronic states with different spin multiplicities, is ubiquitous and imperative in molecular photochemistry. The manifestation of a triplet manifold in π-conjugated chromophoric materials has a crucial role in enhancing the efficiency of photofunctional devices. Herein, we explore the triplet-state population in a series of chalcogen-annulated perylene bisimides (O-PBI, S-PBI, and Se-PBI), where the selenium-annulated PBI (Se-PBI) exhibits a near-quantitative triplet quantum yield ( = 94 ± 1%). Annulation of Se in the PBI core results in a drastic decrease in the fluorescence quantum yield ( = 1.5 ± 0.2%) compared to the bare PBI ( = 97.0 ± 1%), indicating the possibility of an efficient non-radiative decay pathway in the Se-PBI motif. Femtosecond and nanosecond transient absorption measurements unambiguously confirmed the ultrafast triplet population in Se-PBI with an ISC rate constant of = 2.39 × 1010 s–1 and the triplet-state decay to the ground state with a time constant of 3.78 μs. A theoretically calculated spin–orbit coupling constant (VSOC) of 122.4 cm–1 employing the SA-CASSCF/NEVPT2 method has rationalized the excited-state dynamics of Se-PBI. By virtue of the poor SOC between the singlet and triplet states, we observed a partial triplet population in S-PBI, whereas ISC is negligible in O-PBI. We demonstrate an increase in the spin–orbit coupling constant ( ≪ < ) and rate constant of ISC ( ≪ < ) across the series of chalcogen-annulated PBIs (O-PBI, S-PBI, and Se-PBI). The heavier chalcogenide PBI (Se-PBI) thus adds to the array of potential organic photoactive materials for the design of efficient solar energy conversion devices.
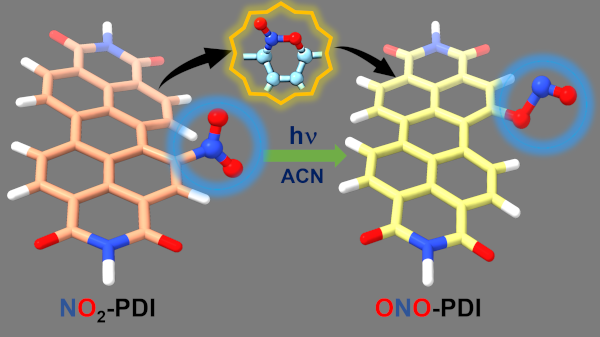
Vibrant excited-state dynamics, in conjunction with distinctive photochemistry has established nitrated-polycyclic aromatic hydrocarbons as an exhilarating class of organic compounds. Herein, we report the atypical photorearrangement of nitro-perylenediimide (NO2-PDI) to nitrito-perylenediimide (ONO-PDI), giving rise to linkage isomers in polar aprotic acetonitrile solvent triggered by visible-light excitation. ONO-PDI has been isolated and unambiguously characterized using standard spectroscopic, spectrometric and elemental composition techniques. Although nitritoaromatic compounds are conventionally considered as crucial intermediates in the photodissociation of nitroaromatics, the experimental evidences for the same are not observed heretofore. Ultrafast transient absorption spectroscopy equipped with computational investigations revealed the prominence of a conformationally relaxed singlet excited-state (S_1^CR) of NO2-PDI in the photoisomerization pathway. Theoretical transition state (TS) analysis displayed the presence of a 6-membered cyclic TS, pivotal in connecting the S_1^CR state to the photoproduct state. This article addresses the prevailing knowledge gaps in the field of organic linkage isomers and provides a comprehensive understanding of the unprecedented photoisomerization mechanism operating in NO2-PDI.
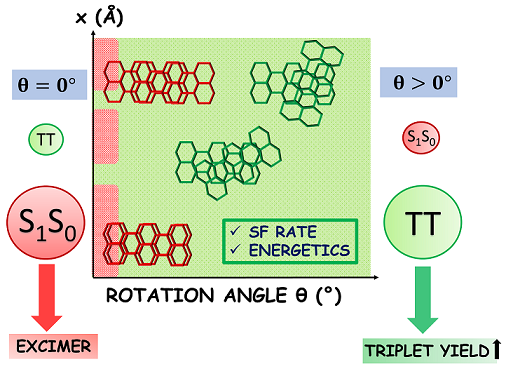
Singlet fission (SF) is a spin-allowed, exciton-multiplying phenomenon that can be utilized to improve the efficiency of organic solar cells. It is well-understood that SF is sensitive to the local crystal morphology and an appropriately balanced coupling is essential to facilitate efficient SF. In this study, we show how the interchromophoric rotation selectively modulates the interaction between the monomer frontier molecular orbitals, promoting both fast and exothermal SF. We evaluate the effective electronic coupling for SF (VSF), the square of which is proportional to the SF rate, and the effective energies of the Frenkel exciton (FE/S1S0) and triplet pair exciton (TT) in a terrylene dimer model. Optimal interplanar rotation of the chromophoric moieties in slip-stacked arrangements pulls the effective energy of the TT state below that of the FE state. Consequently, SF is favored over competing pathways such as excimer formation, thereby enhancing the overall triplet yield. This work represents a step towards improvising the molecular design guidelines for SF and understanding the importance of interchromophoric rotation over the conventional slip-stacked arrangements for achieving favorable intermolecular electronic coupling towards efficient SF.
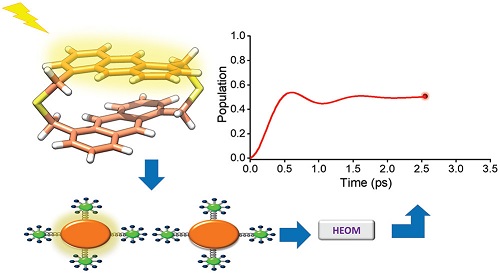
The purpose of this study is to investigate the role of a structured immediate phonon environment in determining the exciton dynamics and the possibility of using it as an optimal design element. Through the case study of dithia-anthracenophane, a bichromophore using the Hierarchical Equations Of Motion formalism, we show that the experimentally observed coherent exciton dynamics can be reproduced only by considering the actual structure of the phonon environment. While the slow dephasing of quantum coherence in dithia-anthracenophane can be attributed to strong vibronic coupling to high-frequency modes, vibronic quenching is the source of long oscillation periods in population transfer. This study sheds light on the crucial role of the structure of the immediate phonon environment in determining the exciton dynamics. We conclude by proposing some design principles for sustaining long-lived coherence in molecular systems.

Nature has been at the forefront of optimizing efficient light energy conversion and energy transduction models in the photosynthetic complex. Inspired by nature, underpinning mechanistic aspects of excited-state energy-transfer processes in assemblies of chromophoric systems, perovskites, and semiconductor materials are of great interest in energy research. However, a comprehensive understanding of the excited-state energy transfer is quintessential for practical implications in photovoltaics and solar fuel generation, which may meet the world’s rising energy need. This virtual issue is a collection of recent research on excited-state energy transfer for light energy conversion published in ACS Energy Letters.
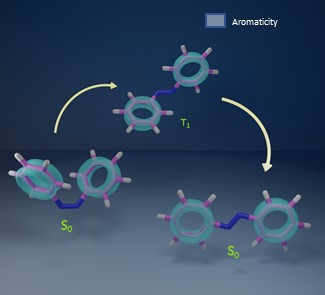
The implication of the potential concept of aromaticity in the relaxed lowest triplet state of azobenzene, an efficient molecular switch, using elementary aromaticity indices based on magnetic, electronic, and geometric criteria has been discussed. Azobenzene exhibits a major Hückel aromatic character retained in the diradical lowest relaxed triplet state (T1) by virtue of a twisted geometry with partial delocalization of unpaired electrons in the perpendicular p-orbitals of two nitrogen atoms to the corresponding phenyl rings. The computational analysis has been expanded further to stilbene and N-diphenylmethanimine for an extensive understanding of the effect of closed-shell Hückel aromaticity in double-bond-linked phenyl rings. Our analysis concluded that stilbene has Hückel aromatic character in the relaxed T1 state and N-diphenylmethanimine has a considerable Hückel aromaticity in the phenyl ring near the carbon atom while a paramount Baird aromaticity in the phenyl ring near the nitrogen atom of the C=N double bond. The results reveal the application of excited-state aromaticity as a general tool for the design of molecular switches.

Despite its identification in the early 19th century, the fundamental understanding of halogen bonding continues to hold immense interest in crystal engineering and supramolecular chemistry. The development of X-ray based diffraction techniques and state-of-the-art theoretical methods has contributed to the significant understanding of this topic in recent years. This Virtual Issue highlights recent publications from Crystal Growth & Design that describe research on halogen bonding. The papers selected include fundamental aspects of halogen bonding and the pivotal role of halogen bonding in modulating the packing in crystalline solids, cocrystallization, supramolecular self-assembly, molecular recognition, and chirality switching.

Architecting unique supramolecular structures requires robust and reproducible supramolecular synthons. Noncovalent halogen bonding offers rich crystal packing possibilities through diverse synthons and thus constitutes a useful structural domain in crystal groups. The variations in halogen synthon patterns with bromine substitution was explored by means of crystal packing analyses of a series of bromine-substituted o-xylenes. We report an unprecedented giant cyclic supramolecular Br8 synthon with a fused triangular prism-like geometry formed exclusively through hypervalent Br···Br interactions in the crystal structure of α,α,α′,α′-tetrabromo-o-xylene. A new polymorph of the compound α,α,α′,α′-tetrabromo-o-xylene with a Br4 tetrahedral synthon was isolated and characterized by single-crystal X-ray diffraction. Additionally, a rectangular Br4 synthon was found to direct the crystal packing in α,α,α′-tribromo-o-xylene. Lines of evidence for the noncovalent intermolecular Br···Br interactions rendering the Br8 and the Br4 synthons were quantitatively obtained by Bader’s quantum theory of atoms in molecules (QTAIM). Noncovalent interaction index isosurfaces exposed the intramolecular interactions, which were not evident from QTAIM. Penda’s interacting quantum atom energy partitioning provided insight into the Br···Br interaction energy in terms of electrostatic and exchange–correlation components. Despite the presence of σ holes as characterized by electrostatic potential analysis, the Br···Br interactions constituting the Br8 and Br4 synthons are stabilized through nonelectrostatic components. The current study intends to reinforce the potential of nonelectrostatic weak halogen···halogen contacts to give rise to a wide variety of synthon patterns and develop solid supramolecular assemblies that find applications in numerous fields of research.
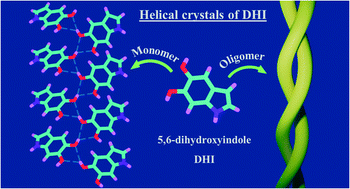
Eumelanin, a naturally occurring group of heterogeneous polymer/aggregate providing photoprotection of living organisms consists of 5,6-dihydroxyindole (DHI) and 5,6-dihydroxyindole-2-carboxylic acid (DHICA) building blocks. Despite their prevalence in the animal world, the structure and therefore, the mechanism behind the photoprotective broadband absorption and non-radiative decay of eumelanin remain largely unknown. As a small step towards solving the incessant mystery, DHI is crystallized in a non-protic solvent environment to render DHI crystals having a helical packing motif. The present approach reflects the solitary directional effect of hydrogen bonds between the DHI chromophores for generating the crystalline assembly and filters out any such involvement of the surrounding solvent environment. The DHI single crystals having an atypical chiral packing motif (P212121 Sohncke space group), incorporate enantiomeric zig-zag helical stacks arranged in herringbone fashion with respect to each other. Each of the zig-zag helical stacks originate from a bifurcated hydrogen bonding interaction between the hydroxyl substituents in adjacent DHI chromophores which act as the backbone structure for the helical assembly. Fragment-based excited state analysis performed on the DHI crystalline assembly demonstrates exciton delocalization along the DHI units that connect each enantiomeric helical stack while, within each stack, the excitons remain localized. Fascinatingly, over the time evolution for generation of single-crystals of DHI-monomer, mesoscopic double-helical crystals are formed, possibly attributed to the presence of covalently connected DHI trimers in the chloroform solution. The oligomeric DHI (in line with the chemical disorder model) along with the characteristic crystalline packing observed for DHI provide insights towards the broadband absorption feature exhibited by the chromophore.
The generation of electron–hole radical pair at the active layer of organic photovoltaics through symmetry-breaking charge separation (SB-CS) has a crucial role in enhancing open-circuit voltage (Voc) and thereby increasing power conversion efficiency. Since the SB-CS materials achieve intramolecular charge separation with a negligible energetic driving force and decelerated charge recombination (CR) rate, SB-CS has been subjected to extensive experimental and theoretical studies. This Focus Review assesses the fundamentals of photosynthetic reaction centers, especially the “special pair”, and discusses how covalent control over the geometric arrangement, surrounding dielectric medium, and substitutions on multichromophoric perylenediimide architecture affects the energy landscape of SB-CS and CR. We systematically summarize the kinetically favored undesirable radiative and non-radiative deactivation channels of SB-CS and CR processes on diverse chromophoric arrangements. Here, we suggest new rational design principles to fine-tune the electron-transfer dynamics at the molecular level to improve the performance of light–energy conversion devices.
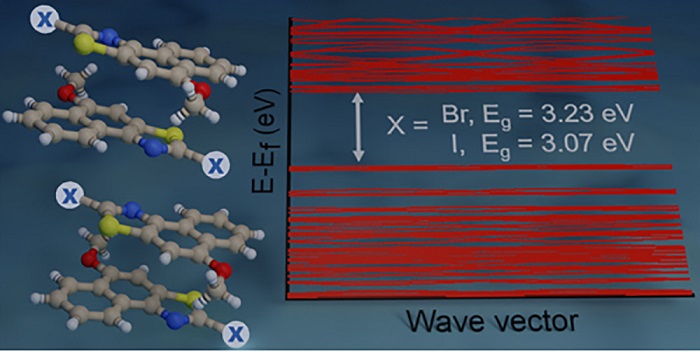
We systematically altered the molecular structure of 5-methoxynaphtho[1,2-d]thiazole (NTH) by replacing the terminal hydrogen atom with halogens (Cl, Br, I) to study the effect of single-atom substitution in modulating the crystal packing and optical band gap. The parent compound (NTH) and Br and I derivatives of NTH crystallized in the same space group, wherein only Br- and I-substituted molecular crystals displayed isostructural interaction topologies. The crystal-packing similarities in structurally equivalent motifs were established using the numerical descriptors isostructurality (Is) and cell similarity (π) indices. An energy framework analysis was implemented to obtain a qualitative picture of the 3D topology displaying the predominant interactions in supramolecular architectures of the NTH derivatives. A decrease in optical band gap from 3.48 to 3.07 eV was observed with an increasing atomic number of halogens in NTH derivatives, signifying the direct role of halogen atoms in the electronic properties of organic crystals. The reduction of the optical band gap in 5-methoxynaphtho[1,2-d]thiazole derivatives was visualized from the band structure and projected density of states obtained by employing DFT calculations. The outcome suggests the potential of halogenation in tailoring the optoelectronic properties of organic functional materials.

Ultrafast charge separation is important for a variety of applications, such as molecular electronics, solar energy conversion, and optical switching. The development of new ultrafast techniques to study charge separation has seen an enormous rise in the number of research articles on this topic in recent years. However, in spite of this progress, questions still remain about what controls the rates of charge transfer and separation in different systems. This Virtual Issue (VI) highlights recent publications from The Journal of Physical Chemistry C that describe research on ultrafast charge separation. The systems studied include organic molecular junctions, polymeric semiconductors, bulk heterojunctions, nanoparticles, and quantum dots.
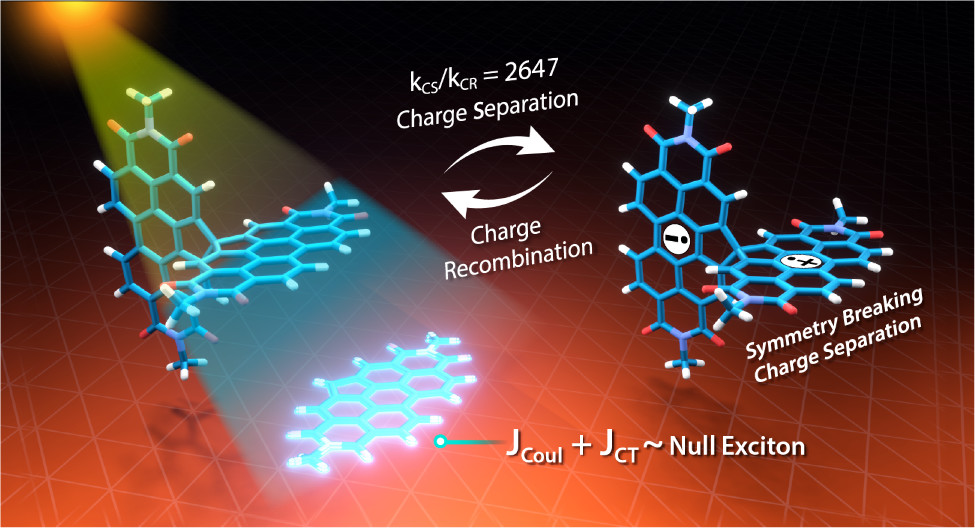
A comprehensive understanding of the structure–property relationships in multichromophoric architectures has pushed the limits for developing robust photosynthetic mimics and molecular photovoltaics. The elusive phenomenon of null exciton splitting has gathered immense attention in recent years owing to the occurrence in unique chromophoric architectures and consequent emergent properties. Herein, we unveil the hitherto unobserved null exciton coupling assisted highly efficient photoinduced symmetry-breaking charge separation (SB-CS) in a Greek cross (+)-oriented spiro-conjugated perylenediimide dimer (Sp-PDI2). Quantum chemical calculations have rationalized the infrequent manifestation of null exciton coupling behavior in Sp-PDI2. Negligible contribution of long-range Coulombic and short-range charge-transfer mediated coupling renders a monomer-like spectroscopic signature for Sp-PDI2 in toluene. The Greek cross (+)-arranged Sp-PDI2 possesses a selective hole-transfer coupling, facilitating the ultrafast dissociation of null excitons and evolution of the charge-separated state in polar solvents. Radical cationic and anionic spectroscopic signatures were characterized by employing femtosecond transient absorption spectroscopy. The substantial hole transfer electronic coupling and lower activation energy barrier of Sp-PDI2 accelerated the charge separation rate. The rate of charge recombination (CR) markedly decelerated due to falling into the inverted region of the Marcus parabola, where the driving force of CR is larger than the total reorganization energy for CR. Hence, the ratio of the rates for SB-CS over CR of Sp-PDI2 exhibited an unprecedently high value of 2647 in acetonitrile. The current study provides impeccable evidence for the role of selective charge filtering in governing efficient SB-CS and thereby novel insights towards the design of biomimics and advanced functional materials.

Inspired by the high photoconversion efficiency observed in natural light-harvesting systems, the hierarchical organization of molecular building blocks has gained impetus in the past few decades. Particularly, the molecular arrangement and packing in the active layer of organic solar cells (OSCs) have garnered significant attention due to the decisive role of the nature of donor/acceptor (D/A) heterojunctions in charge carrier generation and ultimately the power conversion efficiency. This review focuses on the recent developments in emergent optoelectronic properties exhibited by self-sorted donor-on-donor/acceptor-on-acceptor arrangement of covalently linked D–A systems, highlighting the ultrafast excited state dynamics of charge transfer and transport. Segregated organization of donors and acceptors promotes the delocalization of photoinduced charges among the stacks, engendering an enhanced charge separation lifetime and percolation pathways with ambipolar conductivity and charge carrier yield. Covalently linking donors and acceptors ensure a sufficient D–A interface and interchromophoric electronic coupling as required for faster charge separation while providing better control over their supramolecular assemblies. The design strategies to attain D–A conjugate assemblies with optimal charge carrier generation efficiency, the scope of their application compared to state-of-the-art OSCs, current challenges, and future opportunities are discussed in the review. An integrated overview of rational design approaches derived from the comprehension of underlying photoinduced processes can pave the way toward superior optoelectronic devices and bring in new possibilities to the avenue of functional supramolecular architectures.
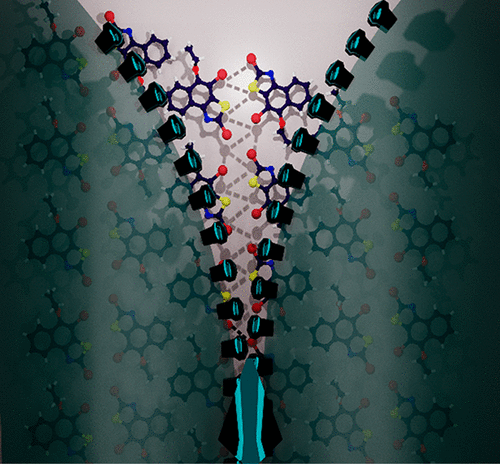
Unprecedented multimodal weak interactions are quintessential to create novel supramolecular topologies. Among the plethora of weak interactions, halogen–halogen (X···X) interactions offer innovative possibilities for the design of multidimensional scaffolds. Herein, we chronicle the state-of-the-art 1D, 2D, and 3D zipper motifs steered by distinct interhalogen interactions, revealing the potential of halogen bonding in engineering biomimetic molecular assemblies. Recurring units of Br4 synthon, framed by type I and type II atom efficient X···X interactions in a dibromonaphthathiazole derivative, 2,4-dibromo-5-ethoxynaphtho[1,2-d]thiazole (NTB2), forges the molecular zipper. On the basis of the semiclassical Marcus theory of charge transport, the NTB2 zipper assembly displays selective electron transport along the type II X···X bonded direction. Band structure analysis classified the crystalline NTB2 as a wide band gap semiconductor with a band gap of 2.80 eV, respectively. The robustness of the X···X mediated zipper motif opens up new avenues in the development of advanced functional materials.
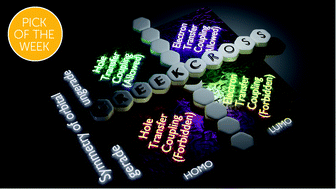
The topology of frontier molecular orbitals (FMOs) induces highly sensitive charge transfer coupling with variation in the intermolecular arrangement. A consistent optoelectronic property correlated to a specific aggregate architecture independent of the nature of the monomer is a rare phenomenon. Our theoretical investigation on stacked dimeric systems of linear [n]acenes (n = 2–5) and selected non-linear acenes with a D2h point group reveals that the Greek cross (+) stacked orientation, irrespective of the molecular candidate, exhibits mutually exclusive hole and electron transfer couplings. The deactivation of either hole or electron transfer coupling is a consequence of the zero inter-orbital overlap between the highest occupied molecular orbitals (HOMOs) or lowest unoccupied molecular orbitals (LUMOs) of the monomers possessing gerade symmetry. In the Greek cross (+) stacked alignment, the (4n + 2) π-electronic acene systems with an odd number of benzenoids exhibit exclusive electron transfer coupling, while the even numbered acenes exhibit selective hole transfer coupling. The trend is reversed for representative 4n π-electronic acene systems. The effect of mutually exclusive charge transfer coupling in the hopping regime of charge transport was evaluated using semiclassical Marcus theory, and selective charge carrier mobility was exhibited by the Greek cross (+) stacks of the considered acene candidates. Additionally, the characteristic charge transfer coupling of the orthogonal acene stacks resulted in negligible short-range exciton coupling, inciting null exciton splitting at short interplanar distances. Engineering chromophores in precise angular orientations ensuring characteristic emergent properties can have tremendous potential in the rational design of advanced optoelectronic materials.
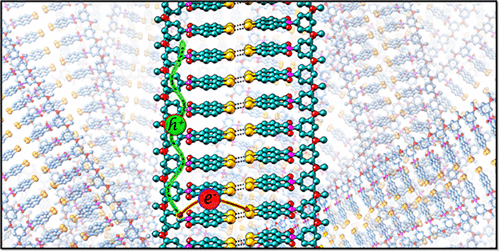
The sophisticated, yet ingenious, supramolecular architectures in nature have often inspired the design of synthetic molecular frameworks mimicking the efficacious emergent properties nurtured by these systems. Herein, the unique crystalline assembly of a dibromonaphthalimide derivative, 1,8-dibromonaphthalene(3,5-dimethoxyphenyl)imide (NIBr2OMe), forming base-pair-like dimers via a stabilizing parallelogram-type Br4 synthon, that further slip-stack to form segregated donor-acceptor arrays, is reported. The peculiar arrangement of the covalently linked donor-acceptor (D-A) moieties with HOMO/LUMO localized on the donor/acceptor part and the peri-peri halogen-halogen interactions imparts higher hole and electron transfer couplings for stacked and halogen–halogen bonded dimers of NIBr2OMe, respectively. The theoretical calculation of anisotropic mobility displayed orthogonal trajectories for maximal hole and electron transport along the slip-stacked and halogen–halogen bonded edge-to-edge directions, respectively. Thus, the unnarrated crucial role of interhalogen interactions in modulating intermolecular electronic couplings and hence the directionality of charge transport is revealed. The study is the first indication for the pre-proposed orthogonal electron and hole transport character in a crystalline organic donor-acceptor system providing novel strategies toward designing archetypical organic materials with charge carrier transport in predetermined trajectories for advanced optoelectronic applications.

Null aggregates are elusive, emergent class of molecular assembly categorized as spectroscopically uncoupled molecules. Orthogonally stacked chromophoric arrays are considered as a highlighted architecture for null aggregates. Herein, we unveil the null exciton character in a series of crystalline Greek cross (+) assembly of 6,13-bisaryl substituted pentacene derivatives. Quantum chemical computations suggest that synergistic perpendicular orientation and significant inter-chromophoric separations realize, negligible long-range Coulombic and short-range charge transfer mediated couplings in the null aggregate. The Greek cross (+) orientation of pentacene dimers exhibit a selectively higher electron transfer coupling with near-zero hole transfer coupling and thereby contribute to the lowering of charge transfer mediated coupling even at shorter inter-chromophoric distances. Additional investigations on the nature of excitonic states of pentacene dimers proved that any deviation from 90° cross-stacked orientation results in the emergence of delocalized Frenkel/mixed Frenkel-CT character and the consequent loss of null exciton/monomer-like properties. The retention of exciton isolation even at short range coupling regime reassures the universality of null excitonic character in perpendicularly cross-stacked pentacene systems. The null-excitonic character was experimentally verified by the observation of similar spectral characteristics in the crystalline and monomeric solution state for 6,13-bisaryl substituted pentacene derivatives. Partitioned influence of aryl and pentacene fragments on interchromophoric noncovalent interactions and photophysical properties respectively resulted in the emergence of pentacene centric Kasha’s ideal null exciton, providing novel in-sights towards the design strategies for cross-stacked chromophoric assemblies. Identifying Greek cross-stacked architecture mediated null excitons with charge filtering phenomenon for the first time in the ever-versatile pentacene chromophoric systems can offer an extensive ground for the engineering of functional materials with advanced optoelectronic properties.
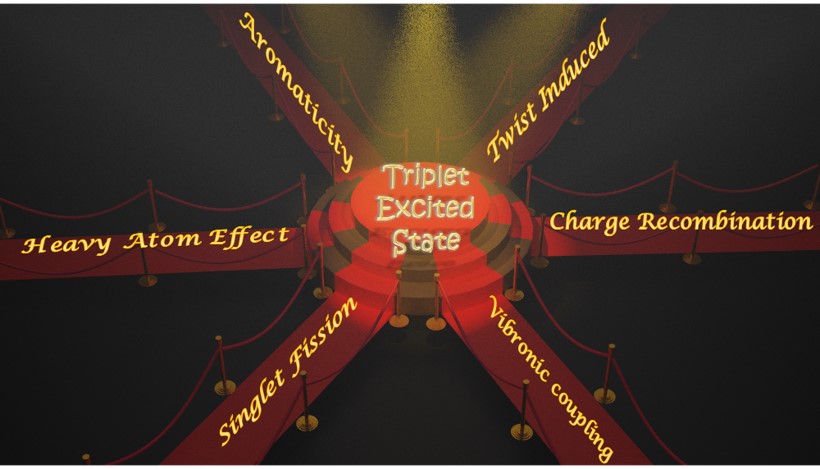
Over the last several decades, exploring the pathways to access the triplet excited states of organic chromophores has been a stimulating area of research. Among the numerous photoinduced processes in organic chromophores, analysis of intersystem crossing (ISC) dynamics has received immense attention. The ISC process involves a spin-forbidden horizontal transition from an excited singlet state to a higher vibrational level of the isoenergetic triplet state. Generally, ISC necessitates a strong driving force from efficient spin–orbit coupling (SOC) between the singlet and triplet wavefunctions. The magnitude of SOC can be tuned by the substituent groups (e.g. heavy atoms, carbonyl moieties) or by the out-of-plane vibrational modes in the chromophores. Besides the SOC induced ISC pathway, triplet excited states are also realised in organic chromophores through singlet fission or via charge recombination. Accessing the triplet manifold in π-conjugated systems would also include a possible evolution to more aromatically stable configurations in the excited states, an emerging area that needs attention. In the aforesaid mechanisms, the molecular architecture and/or packing arrangement of the chromophores are vital for the effective population of triplet states. We, herein, present a collection of synthetic, spectroscopic and theoretical investigations that provide insights into the diverse pathways to access triplet excited states in organic chromophores. We believe this tutorial review would prove beneficial for researchers to achieve triplet excited states of organic chromophores for numerous biochemical and optoelectronic applications.
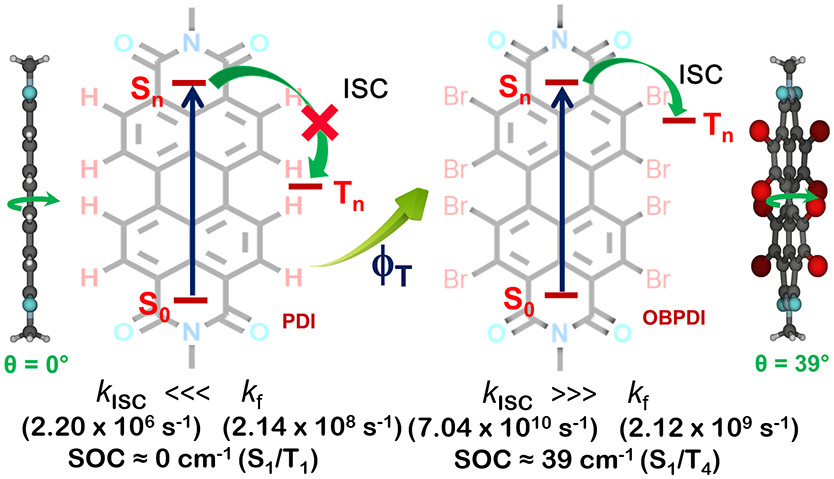
Perylenediimide (PDI) derivatives are essential organic semiconductor materials in a variety of photofunctional devices. By virtue of the large energy gap between the singlet and triplet excited states (ΔEST = 1.1 eV), augmentation of the triplet state population in monomeric PDI is a challenging task. We report the metal atom-free approach in engendering a near-quantitative triplet yield in perbromoperylenediimide/octabromoperylenediimide (OBPDI), absorbing in the visible region of the electromagnetic spectrum. Perbromination of PDI causes significant out-of-plane distortion (θ = 39°) in the aromatic core of OBPDI as compared to the planar PDI (θ = 0°). A substantial decrease (ΔE0red = 0.377 V) in the reduction potential of OBPDI, E1/2(OBPDI/OBPDI·–) = −0.170 V, when compared to the reduction potential, E1/2 (PDI/PDI·–) = −0.547 V, of bare PDI makes OBPDI a promising electron acceptor. As a consequence of incorporating eight bromine atoms, the fluorescence quantum yield of a bare PDI chromophore (ϕf = 97 ± 1%; τf = 4.54 ns) decreases to a very low value in OBPDI (ϕf = 3 ± 1%; τf = 13.78 ps). Femtosecond transient absorption measurements of OBPDI reveal intersystem crossing (ISC) occurring at an ultrafast time scale (τISC = 14.20 ps), leading to a near-quantitative triplet population (ϕT = 97 ± 1%). Theoretical investigations performed to decode the excited state dynamics in OBPDI propose that (i) cumulative addition of eight bromine atoms enhances the magnitude of spin–orbit coupling (SOC) and (ii) twist on the perylene core moderately reduces the energy gap between the singlet–triplet states. Understanding the structural alterations that control the electronic parameters in accessing the triplet excited states of organic chromophores, like PDI, can lead to the design and fabrication of efficient optoelectronic devices and energy storage materials.
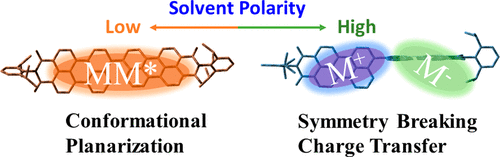
Efficient photoinduced charge separation in artificial multichromophoric architectures relies on two critical factors, (i) electronic coupling and (ii) solvation. While the coherent exciton interactions delocalize the excitation energy among molecules, the solvation-dependent dynamical disorder tends to localize it. Local environments such as solvent polarity/dielectric environments exhibit profound effect on mediating the excited-state relaxation dynamics via specific electronic/geometric changes in chromophore multimers. Herein, a comprehensive account of the solvent governed distinct exciton coupling and symmetry breaking charge separation in a near-orthogonal perylenimide dimer (PP) is presented employing steady-state, femtosecond transient absorption measurements and quantum chemical calculations. Steady-state absorption measurements of the PP dimer reveal apparent electronic coupling between the two monomeric units, wherein the fluorescence measurements reveal a strong fluorescence character in nonpolar solvent, but a significantly quenched state is observed in polar solvent. Ultrafast transient absorption measurements reveal that the fluorescence quenching in polar solvent arises from a photoinduced symmetry-breaking charge transfer (SBCT) process and a subsequent population of the charge-separated radical ion-pair state. Contrastingly, in nonpolar solvent, the charge transfer is endothermic and energetically not feasible. Manifestly, the dimer in nonpolar solvent undergoes a conformational planarization within 20 ps accompanied by excitation delocalization over the two identical monomers in the lowest excited singlet state as evident from the dominant stimulated emission (around 580 nm) and the excited-state absorption (around 740 nm) in the femtosecond transient absorption spectra. Observed solvent-mediated selective control on the excited-state relaxation pathways in the near-orthogonal PP dimer can help shed light on the mechanisms of energy/charge transfer in molecular systems and guide the design of novel high-performance photovoltaic materials..
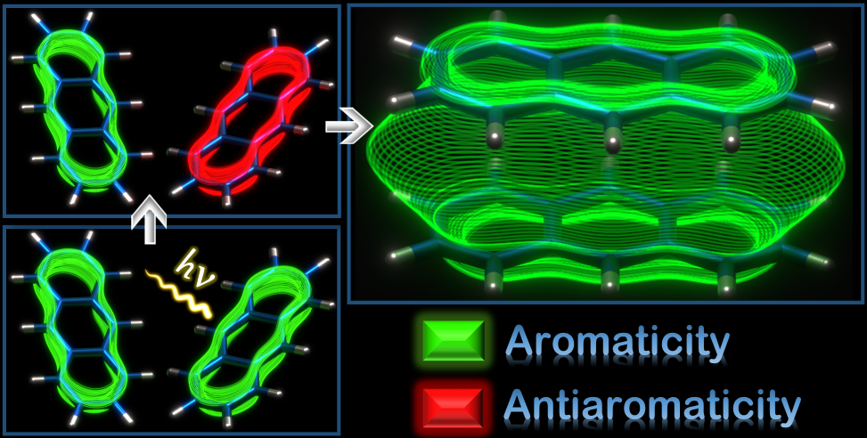
Aromaticity, though widely used to delineate diverse photochemical phenomena, remains to be examined in excimers, a fundamental and extensively studied entity in the excited states. Herein, the first theoretical evidence for excited state through-space aromatic character in triplet state (T1) excimers of benzene, naphthalene and anthracene is reported using multiple aromaticity descriptors based on magnetic, electronic and geometric criteria. Calculated chemical shifts and induced current densities manifest the presence of transannular π-electronic currents in the excimers. The results open up enormous research potential from exploring the possibility of through-space aromatic character in singlet excimers to its possible implications in photoexcited state processes of aromatic supramolecular systems.
Self-assembly of chiral organic chromophores garners huge significance owing to the abundance of supramolecular chirality found in natural systems. We report an interdigitated molecular organization involving axially chiral twisted octabrominated perylenediimide (OBPDI) transferring chiral sense to achiral aromatic moieties. The two‐component crystalline architectures of OBPDI and electron rich aromatic units were facilitated through π-hole•••π based donor-acceptor interactions and the charge transfer characteristics in the ground and excited states of OBPDI cocrystals were established through spectroscopic and theoretical techniques. The OBPDI cocrystals entailed a remarkable homochiral segregation of P and M enantiomers of both the molecular entities in the same crystal system to render twisted double racemic architectures. Synergistically engendered cavities with stored chiral information of twisted OBPDI stabilized higher energy P/M enantiomers of trans‐azobenzene through non-covalent interactions.

Spatial noncovalent helical organization of nucleobases in DNA and radial organization of chromophores in natural light-harvesting systems are fascinating yet enigmatic. Understanding the numerous weak interactions that drive the formation of elegant supramolecular architectures in native natural systems and developing bioinspired design strategies have seen a surge of interest in recent decades. Self-assembly of functional chromophores in the crystalline phase is a definitive strategy to identify novel molecule–molecule interactions, in particular, atom–atom interactions, and to understand the synergistic nature of noncovalent interactions that stabilizes the supramolecular organization. This Account narrates our recent efforts in developing desirable supramolecular motifs employing weak interaction-based strategies and our observation of deviations from the common motifs chartered in aromatic systems. Modulation of long-range aromatic interactions through chemical modifications (acylation, benzoylation, haloacylation, and alkylation of chromophores) to attain a preferred stacking (herringbone, lamellar, or columnar) is presented. Particular attention has been given to attaining lamellar or columnar packing possessing potential interchromophoric electronic coupling mediated high charge mobility. Supramolecular arrangements of noncovalently or covalently associated donor–acceptor systems that open up additional possibilities of packing modes (segregated, mixed etc.) are explored. Our persistent efforts yielded distinct twisted-segregated and alternate distichous stacks for the nonparallel covalently linked donor–acceptor systems that favor a long-lived photoinduced charge-separated state. We further move on to discuss the unconventional packing motifs that were identified recently. The highly sought-after Greek cross (+) stacking of chromophores in crystalline phase and an elegant crystalline radial arrangement of chromophores are examined. The Greek cross (+) stacked architecture exhibits monomer-like emission characteristics owing to the absence of exciton coupling across the orthogonally stacked chromophores. Crystalline helical chromophore assembly is yet another emerging motif with far-reaching applications in domains ranging from asymmetric catalysis to chiral smart materials and has been accounted here by citing certain phenomenal examples from literature. Thus, this Account demonstrates that identifying and classifying new structural motifs based on topological aspects, such as interchromophoric orientation (cross) and extended chromophore arrangement in the crystal lattice (radial, helical, etc.), are crucial since such fundamental characteristics dictate the properties emerging out of the corresponding motifs. Encouraged from ours and others’ works, we propose the addition of new aromatic supramolecular structural motifs, namely, cross-stacked, helical, and radial arrangements, in order to expand the classification. We believe that identifying new emergent property-based supramolecular motifs and investigating the methods to achieve the desired motif will eventually have implications in fundamental crystal engineering, supramolecular chemistry, and biomimetic design of functional materials.
A series of π-conjugated tetrabenzoacenes (TBA), including nitrogen (un)doped derivatives are computationally evaluated to comprehend the correlation between intrinsic structural arrangements and charge transport characteristics. The central charge transport parameters such as reorganization energy and electronic coupling are individually tuned through peri-substitutions, core-substitutions and/or π-extension in TBA derivatives. Based on reorganization energies, nitrogen doping impeded the electron transport in TBA analogs owing to significant structural changes associated with the reduction process. Our approach employing mapping of dimeric arrangements of TBA, modulated via long (pitch) and short (roll) axes displacements of the molecular entities, versus charge transfer coupling disclosed potential charge transport regions in addition to the ideal cofacial modes. Charge transport characteristics of molecular packing arrangements of TBA mimicking the different orientations of graphene bilayers were analyzed, providing insights into the possible material applicability of TBA derivatives. The transition from completely aligned graphitic AA packing sequence to slip-stacked AB and AA’ stacking domains revealed a dent in the charge transport map owing to node-antinode interaction of the frontier molecular orbitals. TBA analogs encompassing expanded π-system materialized highly displaced dimeric orientation from AB type packing to occupy a hierarchy favoring high charge transfer coupling than the AB type. Thus, realizing stable interchromophoric arrangements of small organic molecules through chemical or physical techniques to control their charge transporting efficiencies is an indispensable step towards the generation of better organic electronic devices.
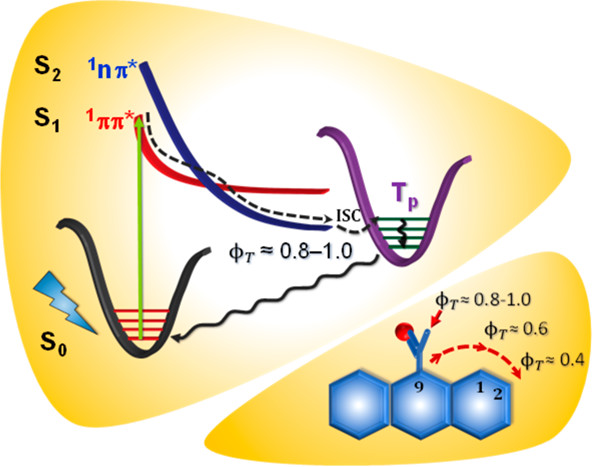
Mapping the primary photochemical dynamics and transient intermediates in functional chromophores is vital for crafting archetypal light-harvesting materials. Although the excited state dynamics in 9-acetylanthracene is well explored, the origin of near-quantitative triplet population and the atypical intersystem crossing (ISC) rate as compared with the regioisomeric analogs (1-/2-acetylanthracene) have rarely been scrutinized. We present a comprehensive account of the photoinduced dynamics in three regioisomeric monoacetylanthracenes using ultrafast transient absorption and quantum chemical calculations. The conjoint experimental and computational investigations suggest that (i) greater stabilization of the 1nπ* relative to 1ππ* state, (ii) dissimilar 1ππ* → 1nπ* crossover barriers, and (iii) the strong spin–orbit coupling (νSO) of the 1nπ* state with the receiver 3ππ* state command the divergent triplet population in 1-/2-/9-acetylanthracenes. A tacit understanding of the subtle structural-alteration-facilitated contrasting ISC dynamics in carbonylated arenes can act as a stepping stone for the evolution of potent photofunctional materials.
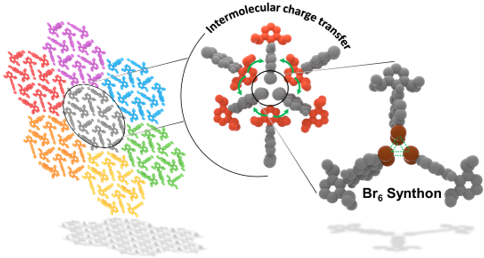
The design of highly efficient supramolecular architectures that mimic competent natural systems requires a comprehensive knowledge of noncovalent interactions. Halogen bonding is an excellent noncovalent interaction that forms halogen-halogen (X2) as well as trihalogen interacting synthons. Herein, we report the first observation of a symmetric radial assembly of chromophores (R-3c space group) composed of a stable hexabromine interacting synthon (Br6) that further push the limits of our understanding on the nature, role and potential of noncovalent halogen bonding. Contrary to the destabilization proposed for Type-I X2 interactions, Br6 synthon possessing Type-I X2 interactions exhibit a stabilizing nature owing to the exchange-correlation component. The radial assembly of chromophores is further strengthened by intermolecular through-space charge transfer interaction. Br6 synthon driven 3-fold symmetric radial assembly render a lattice structure that reminisces the chromophoric arrangement in the light harvesting system 2 of purple bacteria.
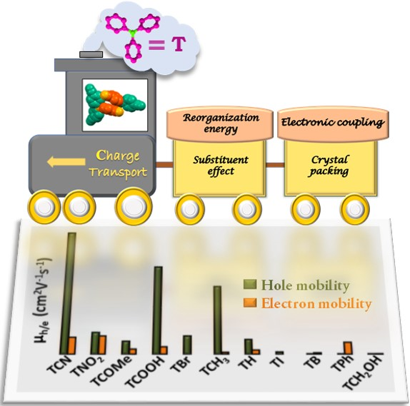
A collection of para‐substituted propeller-shaped triphenylamine (TPA) derivatives have been computationally investigated for the charge transport characteristics exhibited by the derivatives using the Marcus‐Hush formalism. The various substituents chosen in the current work, having features that range from electron withdrawing to electron donating nature, play a key role in defining the reorganization energy and electronic coupling properties of the TPA derivatives. TPA moiety is expected to possess weak electronic coupling on the basis of poor orbital overlap upon aggregation owing to the restriction imposed by the propeller shape of the triphenylamine core. However, the substituent groups attached to the TPA core can significantly dictate the crystal packing motif of the TPA derivatives, wherein the variety of noncovalent intermolecular interactions subsequently generated drive the packing arrangement and influence the electronic coupling between the neighbouring orbitals. The intermolecular interactions in the crystalline architecture of TPA derivatives were probed using Hirshfeld and QTAIM techniques. Furthermore, SAPT analysis of the TPA analogues has revealed that a periodic arrangement of energetically stable dimers having significant electronic coupling is essential in order to contribute a high charge carrier mobility to the overall crystal.
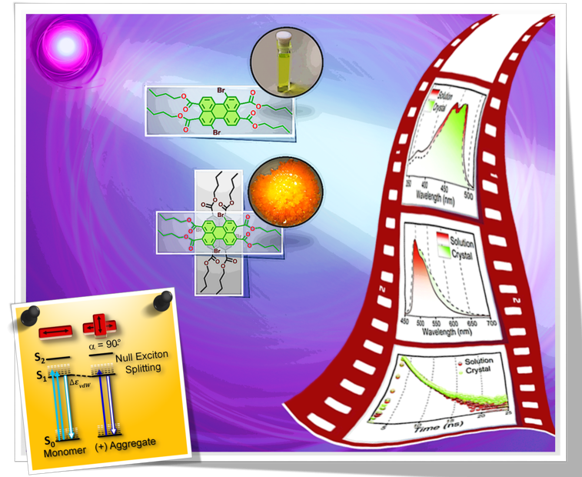
Exciton interactions in molecular aggregates play a crucial role in tailoring the optical behavior of π-conjugated materials. Though vital for optoelectronic applications, ideal Greek cross-dipole (α = 90°) stacking of chromophores remains elusive. We report a novel Greek cross (+) assembly of 1,7-dibromoperylene-3,4,9,10-tetracarboxylic tetrabutylester (PTE-Br2) which exhibits null exciton coupling mediated monomer-like optical characteristics in crystalline state. Contrastingly, nonzero exciton coupling in X-type (α = 70.2°, PTE-Br0) and J-type (α = 0°, θ = 48.4°, PTE-Br4) assemblies render perturbed optical properties. Additionally, the semi-classical Marcus theory of charge-transfer rates predicts a selective hole transport phenomenon in the orthogonally stacked PTE-Br2. Precise rotation angle dependent optoelectronic properties in crystalline PTE-Br2 can have consequences in the rational design of novel π-conjugated materials for photonic and molecular electronic applications.
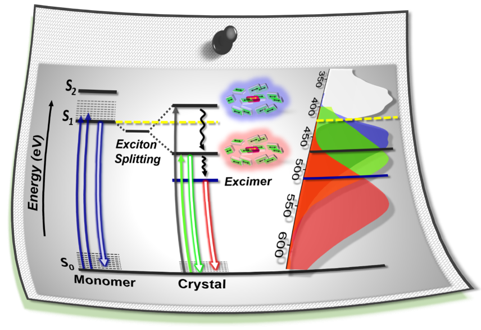
We report a conjoint theoretical and experimental investigation on the role of concerted long‐ (dipole coupling) and short‐range (orbital overlap mediated excimer) electronic interactions in modulating the emission of six crystalline acetylanthracenes (1‐3). Friedel‐Crafts acylation of anthracene rendered crystalline acetylanthracenes with discrete close‐packing, varied orbital overlap, and resultant distinct emission (blue‐green‐yellow) from cooperative excimer and dipole coupling. Time‐resolved emission spectral (TRES) studies and Kasha’s exciton theory based quantitative estimation of dipole coupling (mean field approximation) substantiates the exciton dynamics in crystalline 1‐3. Extension of Kasha’s exciton model beyond the traditional nearest‐neighbor approach, and consistent agreement among the computed spectral shifts and TRES temporal components corroborate a holistic approach to decipher the exciton relaxation dynamics in molecular assembly of novel photonic materials.
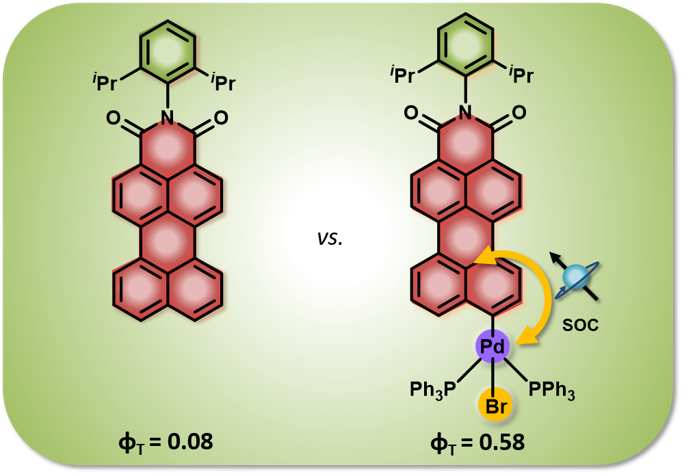
Triplet excited state in organic chromophores is ubiquitously significant owing to its utility in light harvesting and photovoltaic device applications. Herein, we report the enhancement in the triplet character of an innately triplet deficient peryleneimide chromophore via incorporation of a heavy atom. Palladium incorporated perylenemonoimide (PMI-Pd) was synthesized via oxidative addition of PMI-Br into Pd(0) under inert experimental conditions. The structural sanctity of the PMI-Pd and the model derivative PMI was characterized via single crystal X-ray diffraction and the close-packing was examined employing Hirshfeld surface analysis. The steady-state spectroscopic measurements of PMI-Pd in chloroform reveal an apparent perturbation in the UV-Vis absorption, fluorescence emission and lifetime characteristics. A much higher perturbation is observed in the ultrafast photoexcited processes of PMI-Pd in chloroform as envisaged via nanosecond transient absorption (nTA) measurements. The nTA measurements of PMI-Pd in chloroform reveal a significant enhancement in the triplet character of PMI-Pd as compared to the model derivative PMI. Spin-orbit coupling (SOC) mediated triplet enhancement in PMI-Pd suggest heavy atom incorporation as a viable route for accessing the triplet excited states in triplet deficient aromatic chromophores. SOC mediated triplet enhancement in innately triplet deficient organic chromophores can revive the utility of these materials for novel photovoltaic and energy storage applications.
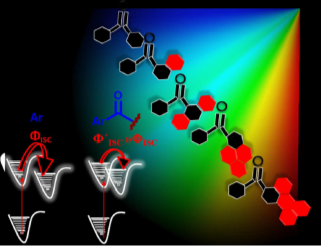
A series of extended π-conjugated benzophenone analogs was synthesized through the facile Lewis-acid catalyzed Friedel-Crafts reaction in order to exploit the integral triplet state properties of benzophenone. Extending the π-conjugated plane of the phenyl ring of benzophenone allowed to tune the excitation wavelength from far-UV end (~260 nm) to the visible spectrum (~446 nm). Compared to benzophenone, significant red-shifts in the absorption (up to 450 nm in solution) with high photostability, were established for the synthesized benzophenone analogs. As evident from the density functional theory calculations, expansion of ring size at the aromatic part in benzophenone analogs induces a decrease in the HOMO-LUMO gap. The considerable extension of electron density to the carbonyl group in the LUMO substantiates the triplet nature associated in the benzophenone analogs. By virtue of the properties of carbonyl functionality, an apparent increase in the triplet quantum yield (ΦT = 5.4 to 87.7 %) was observed for benzophenone analogs when compared to the corresponding bare polyaromatic hydrocarbon. The spin-orbit coupling was computationally estimated for the benzophenone analogs to propose pathways for the observed intersystem crossing process. The plausibility to photoexcite the aromatic-ring-fused benzophenone frameworks for triplet activation in the visible range opens the door for a new class of materials for photonic application.
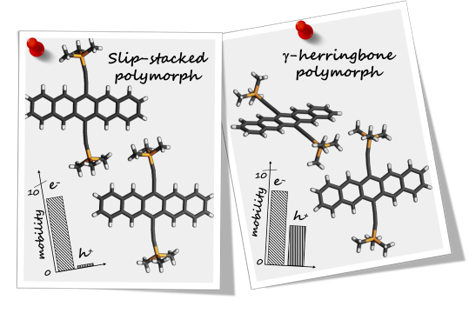
The introduction of the trialkylsilylethynyl group to the acene core is known to predominantly transform the herringbone structure of pentacene to a slip‐stacked packing. However, in this report, we realize the occurrence of an unforeseen polymorph of 6,13‐bis(trimethylsilylethynyl)pentacene (TMS‐pentacene) with an atypical γ‐herringbone packing arrangement. The intermolecular non‐covalent interactions in the γ‐herringbone polymorph are determined from Hirshfeld surface and QTAIM analyses. Furthermore, a comparative SAPT(0) energy decomposition analysis discloses the role of exchange repulsions that govern the molecular packing in the γ‐herringbone polymorph. Moreover, the computationally predicted electronic coupling and anisotropic mobility reveals the possibility of enhanced hole transport (µh=3.7 cm²V‐¹s‐¹) in the γ-herringbone polymorph in contrast to the reported polymorph with the hole mobility of µh=0.1 cm²V-¹s-¹.
Among the various donor-acceptor (D-A) charge transfer cocrystals investigated in the past few decades, tetrathiafulvalene-tetracyanoquinodimethane (F.Q, popularly known as TTF.TCNQ) based cocrystals have fascinated materials chemists owing to its packing and exceptional properties. Here, crystallographic information files of eighteen F.Q based cocrystals were extracted from Cambridge Structural Database and classified into Class 1 (D on D and A on A segregated stacks; F.Q, F1.Q - F6.Q and F.Q1), Class 2 (-A-D-A-D-A-D- mixed stacks; F6a.Q - F11.Q and F.Q2) and Class 3 (-A-D-A-A-D-A-; Class 3a (F12.Q and F13.Q) and -D-D-A-A-D-D-; Class 3b (F14.Q)) systems based on their packing modes. PIXEL calculations revealed that the Q on Q dimer is the energetically most favored dimer in F.Q, the substituents on F capable of forming hydrogen bonding, C...S and other weak intermolecular interactions resulted in the greater stability of F on F dimer for F1.Q - F6.Q (except F2.Q). Band structure of F.Q and F6.Q with high interaction of electronic orbitals between D on D and A on A in segregated stacks were found to be metal-like (band gap, Eg = 0.003 eV) and metallic (overlapping bands in the Fermi level) respectively while the polymorph of F6.Q belonging to Class 2 (F6a.Q) displayed a semiconductor-type band structure (Eg = 0.053 eV). F12.Q of Class 3a exhibited a metal-like band structure (Eg = 0.001 eV).
The photophysics of several mono- and oligonucleotides were investigated in a deep eutectic solvent for the first time. The solvent glyceline, prepared as a 1 : 2 mole ratio mixture of choline chloride and glycerol, was used to study excited-state deactivation in a non-aqueous solvent by the use of steady-state and time-resolved spectroscopy. DNA strands in glyceline retain the secondary structures that are present in aqueous solution to some degree, thus enabling a study of the effects of solvent properties on the excited states of stacked bases and stacked base pairs. The excited-state lifetime of the mononucleotide 5′-AMP in glyceline is 630 fs, or twice as long as in aqueous solution. Even slower relaxation is seen for 5′-TMP in glyceline, and a possible triplet state with a lifetime greater than 3 ns is observed. Circular dichroism spectra show that the single strand (dA)18 and the duplex d(AT)9·d(AT)9 adopt similar structures in glyceline and in aqueous solution. Despite having similar conformations in both solvents, femtosecond transient absorption experiments reveal striking changes in the dynamics. Excited-state decay and vibrational cooling generally take place more slowly in glyceline than in water. Additionally, the fraction of long-lived excited states in both oligonucleotide systems is lower in glyceline than in aqueous solution. For a DNA duplex, water is suggested to favor decay pathways involving intrastrand charge separation, while the deep eutectic solvent favors interstrand deactivation channels involving neutral species. Slower solvation dynamics in the viscous deep eutectic solvent may also play a role. These results demonstrate that the dynamics of excitations in stacked bases and stacked base pairs depend not only on conformation, but are also highly sensitive to the solvent.
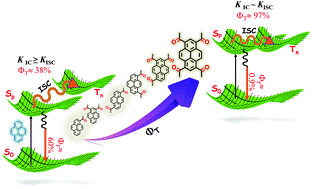
Ultrafast intersystem crossing of carbonylpyrenes in chloroform was investigated by femtosecond pump–probe spectroscopy. When compared to the dominant fluorescence decay pathway in pyrene, carbonyl functionalized pyrenes display near-unity triplet formation upon photoexcitation. The excited singlet state (Sp) undergoes rapid intersystem crossing (kISC) concomitantly with internal conversion (kIC) to lower excited singlet states (Sn) within a timescale of 5–11 ps (1/τ2 = kIC + kISC). Furthermore, intersystem crossing from lower excited singlet states (Sn) proceeds through coupling with receiver triplet states, eventually leading to high triplet quantum yields (ΦT = 97%; tetraacetylpyrene). Followed by internal conversion in the triplet manifolds, phosphorescence decay on a microsecond timescale is observed from the emitter triplet state.
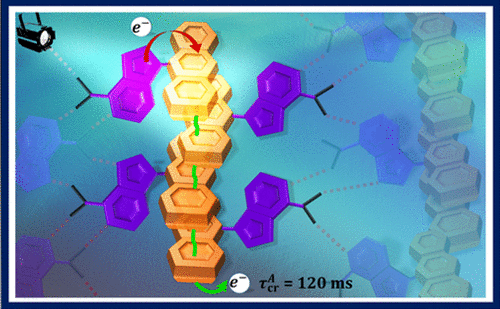
We report a comprehensive analysis of the ultrafast photoexcited-state processes of a nucleobase–arene conjugate, 9-(adenin-9-yl)anthracene (AdAn), in organic solvents. By virtue of the strong base-pairing and π–π interactions, AdAn forms an alternate distichous assembly, wherein the adenines are near-orthogonally flanked to the columnar anthracene-on-anthracene (An–An) stacks. The preliminary photophysical and redox analysis of AdAn in organic solvent reveals a summation of adenine (Ad) and anthracene (An) spectral features with minimal perturbation in the ground-state properties. The frontier molecular orbital analysis suggests favorable charge transfer characteristics in the dyad, which is further corroborated by the negative value of the free energy for electron transfer from 1Ad* to An (ΔGet = −1.25 eV) and Ad to 1An* (ΔGet = −0.12 eV) obtained via Weller analysis. Upon photoexcitation (λex = 266 and 355 nm), self-assembled AdAn reveals the formation of radical ion-pair intermediates (τcrA = 120 ms), which display a solvent polarity dependence as demonstrated via nanosecond and femtosecond transient absorption spectroscopy. The “emergence upon assembly” approach offers segregated trajectories of the charge carriers, i.e. delocalization of the electron through the columnar π-channels of anthracenes and the diffusion of the holes across hydrogen-bonded adenines, resulting in persistent radical ion-pair intermediates. Though the unsolicited photoexcited-state pathways of AdAn lead to undesirable photoproducts, the long-lived charge-separated state from a favorable arrangement of DA stack proposes an elegant DA assembly design for emergent solution processable photofunctional devices.
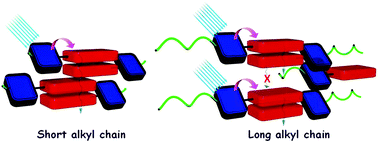
Side-chains at the imidic position of naphthalimide rendered a firm control over (i) the degrees of π–π overlap and (ii) distances between the perylenimide units in a crystalline naphthalimide–perylenimide dyad as determined using single crystal XRD and Hirshfeld surface analyses. Steady-state and time-resolved electronic spectroscopy in addition to DFT calculations revealed a decline in intermolecular excitonic interaction due to interfering alkyl chains.
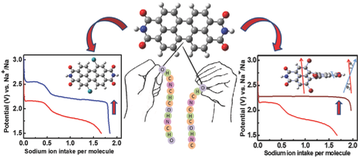
Organic rechargeable batteries gain huge scientific interest owing to the design flexibility and resource renewability of the active materials. However, the low reduction potentials still remain a challenge to compete with the inorganic cathodes. This study demonstrates a simple and efficient approach to tune the redox properties of perylene diimides (PDIs) as high voltage cathodes for organic-based sodium-ion batteries (SIBs). With appropriate electron-withdrawing groups as substituents on perylene diimides, this study shows a remarkable tunability in the discharge potential from 2.1 to 2.6 V versus Na+/Na with a sodium intake of ≈1.6 ions per molecule. Further, this study explores tuning the shape of the voltage profiles by systematically tuning the dihedral angle in the perylene ring and demonstrates a single plateau discharge profile for tetrabromo-substituted perylene diimide (dihedral angles θ1 & θ2 = 38°). Detailed structural analysis and electrochemical studies on substituted PDIs unveil the correlation between molecular structure and voltage profile. The results are promising and offer new avenues to tailor the redox properties of organic electrodes, a step closer toward the realization of greener and sustainable electrochemical storage devices.
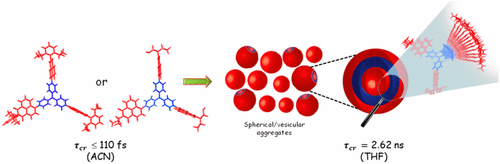
Organic photonic nanostructures, capable of efficient light harvesting and storage, provide new avenues in constructing solution processable solar cells and photovoltaic devices. In this contribution, we demonstrate ca. 104-fold enhancement in the photoinduced charge recombination lifetime (τcra = 2.62 ns) in the aggregated state of donor–acceptor (D–A) dyads and trefoils comprised of triphenylamine and naphthalimide. The D–A dyads and trefoils undergo self-assembly in THF forming spherical/vesicular aggregates dictated by weak co-operative intermolecular interactions in contrast to monomer in CH3CN. Observed long-lived charge transfer intermediates in the aggregated state of triphenylamine-naphthalimide (TN) based conjugates could be attributed to the delocalization of photogenerated charge carriers through D–A stacks. D–A supramolecular architectures thus emerged could serve as promising scaffolds for light harvesting, molecular electronics and photofunctional applications.
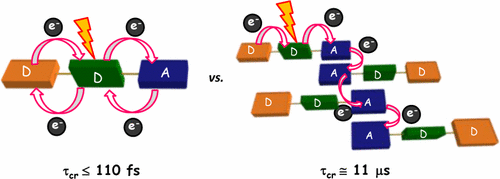
Organic materials which can self-assemble into higher-order superstructures have extensive applications in artificial light-harvesting systems, solution-processable bulk-heterojunction solar cells, and photofunctional devices owing to their unique charge transport properties. In this report, we demonstrate a self-assembled nonsymmetric donor–acceptor triad (TAN) composed of triphenylamine (T), anthracene (A), and naphthalimide (N) units, for achieving long-lived charge separation via aggregation. Steric hindrance imposed by diisopropyl groups of naphthalimide and the propeller-shaped triphenylamine unit obstructs planarization in TAN. The quantum theory of atoms in molecules demonstrated the presence of synergistic C–H···π, C–H···H–C, π–π, and C–O···O–C interactions between the adjacent TAN units in the crystalline state, leading to significant electronic coupling (Hab). The nonplanar geometry of the TAN triad in the monoclinic space group dictates a unique antiparallel arrangement between the adjacent TAN units along the b-axis. Solvent polarity dependent Lippert–Mataga and spin density distribution analyses of TAN established the presence of charge transfer interactions in the molecule. Solvent polarity dependent nanosecond and femtosecond transient absorption measurements of TAN revealed ca. 108-fold enhancement in the lifetime of the charge-separated state in the aggregated TAN in CHCl3 (τcra ≈ 11 μs) and THF (τcra ≈ 11.20 μs) when compared to that in monomeric TAN (τcrm < 110 fs) in CH3CN. Extension in lifetime of the charge-separated state in self-assembled TAN could be due to the synergetic effect induced by delocalization of charge carriers across alternate slipped antiparallel (Hab = 13 meV) and antiparallel dimers (Hab = 10 meV) of TAN and the presence of triplet charge-separated states.
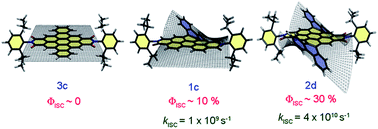
We describe the design, bottom-up synthesis and X-ray single crystal structure of systematically twisted aromatics 1c and 2d for efficient intersystem crossing. Steric congestion at the cove region creates a nonplanar geometry that induces a significant yield of triplet excited states in the electron-poor core-twisted aromatics 1c and 2d. A systematic increase in the number of twisted regions in 1c and 2d results in a concomitant enhancement in the rate and yield of intersystem crossing, monitored using femtosecond and nanosecond transient absorption spectroscopy. Time-resolved absorption spectroscopic measurements display enhanced triplet quantum yields (ΦT = 10 ± 1% for 1c and ΦT = 30 ± 2% for 2d) in the twisted aromatics when compared to a negligible ΦT (<1%) in the planar analog 3c. Twist-induced spin–orbit coupling via activated out-of-plane C–H/C[double bond, length as m-dash]C vibrations can facilitate the formation of triplet excited states in twisted aromatics 1c and 2d, in contrast to the negligible intersystem crossing in the planar analog 3c. The ease of synthesis, high solubility, access to triplet excited states and strong electron affinity make such imide functionalized core-twisted aromatics desirable materials for organic electronics such as solar cells.
Tuning the luminescence properties of solid state materials by controlling the molecular packing in the crystal has played an important role in designing new functional materials. A series of five V-shaped oxydiphthalimides (ODPs) were synthesised. Side-chain engineering has been successfully utilized to systematically tune their luminescence properties in the crystalline state by introducing sterically bulky side-chains at the imidic nitrogen. Fluorescence spectroscopy and single crystal X-ray analyses disclose the effect of conformational twisting and bulkiness of imidic substituents on crystallisation-induced emission enhancement (CIEE). Detailed analysis revealed that the CIEE factor of ODPs shows a linear dependence on the percentage volume of the side-chain.
The crystal packing and solid state photophysical properties of twisted propeller-shaped triphenylamine (T) incorporated aromatic hydrocarbons [ArT where Ar = benzene (Ph), naphthalene (N), anthracene (A), phenanthrene (Phe), pyrene (Py) and perylene (Pe)] are investigated. The qualitative crystal structure and quantum theory of atoms-in-molecules (QTAIM) analyses revealed the dominant role of intermolecular C–H⋯C interactions in governing the three-dimensional arrangement in ArT crystals. Hirshfeld surface analyses revealed that the triphenylamine-substituted arene (ArT) crystals possess a herringbone motif with ρ (%C–H⋯C/%C⋯C) values varying from 10.4 in PeT to 101.2 in AT. A blue shift of ca. 10–28 nm in the emission maxima for PhT, NT, AT, PheT and PyT is observed in the solid state in comparison to that in dichloromethane while the solid state emission maxima is red shifted (ca. 25–71 nm) compared to that in hexane. A 2.40-fold enhancement in the quantum yield of AT is observed in the solid state relative to the solution state (dichloromethane). Solvent polarity dependent absorption and emission measurements of ArT derivatives along with Lippert–Mataga analyses evidenced the presence of charge transfer interactions between Ar and T units. A precise and systematic understanding of the influence of the crystal structure on the photophysical properties is quintessential to achieve highly efficient organic light emitting diodes and other optoelectronic devices in the near future.
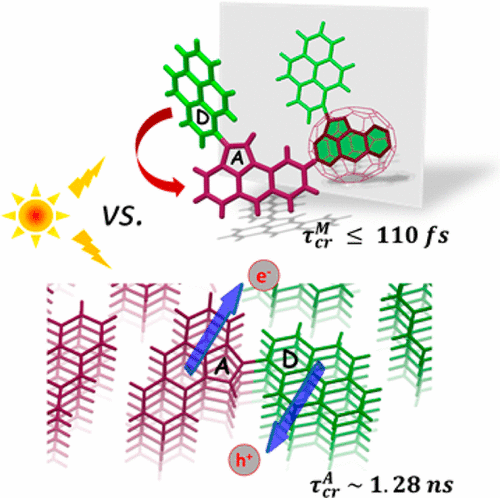
Twisted donor-on-donor and acceptor-on-acceptor bicontinuous assembly in all-carbon pyren-1-ylaceanthrylene (PA) dyad extends the survival time of the photoinduced radical ion-pair intermediates. Aceanthrylene, a functional analog of C70, acts as a versatile electron acceptor owing to its high electron affinity and visible light absorption. Antithetical trajectories of the excitons in the nonparallel π-ways led to persistent radical ion-pair intermediates in aggregated (τcrA ∼ 1.28 ns) vs monomeric (τcrM ≤ 110 fs) PA dyad as observed using femtosecond transient absorption spectroscopy. Marcus theory of charge transfer rates predicts an ambipolar transport characteristic in crystalline PA, thereby endorsing PA as an all-carbon DA hybrid for nonfullerene photovoltaic applications.
The fluorescent probe 2-aminopurine (2Ap) has been used for decades to study local conformational fluctuations in DNA. Steady-state and time-resolved measurements of 2Ap fluorescence have been used to predict specific conformational states through suitable modeling of the quenching of the fluorescence of a 2Ap residue incorporated site-specifically into a DNA strand. The success of this approach has been limited by a lack of understanding of the precise factors responsible for the complex, multiexponential decays observed experimentally. In this study, dinucleotides composed of 2Ap and adenine were studied by the time-correlated single-photon counting technique to investigate the causes of heterogeneous emission kinetics. Contrary to previous reports, we argue that emission from 2Ap that is stacked with a neighboring base contributes negligibly to the emission signals recorded more than 50 ps after excitation, which are instead dominated by emission from unstacked 2Ap. We find that the decay kinetics can be modeled using a continuous lifetime distribution, which arises from the inherent distance dependence of electron transfer rates without the need to postulate a small number of discrete states with decay times derived from multiexponential fits. These results offer a new perspective on the quenching of 2Ap fluorescence and expand the information that can be obtained from experiments.
Stimulated by strongly directional C–I···N noncovalent halogen bonding, π-hole···π and π–π interactions, cocrystals of nonplanar 4-arylated-2,2′-bipyridine (ArB) derivatives with 1,4-diiodo-tetrafluorobenzene (D) were generated which exhibit a promising columnar/lamellar packing arrangement. Hirshfeld surface, quantum theory of atoms in molecules, and electrostatic potential surface analyses were employed to examine the weak intermolecular interactions governing the packing arrangement in ArB crystals and corresponding cocrystals with D (ArB·D). Cocrystals of 4-phenyl-2,2′-bipyridine (PhB) and 4-(naphthalen-1-yl)-2,2′-bipyridine (NaB) with D [PhB·D1, PhB·D2, (NaB)2·D2.5, and (NaB)3·D2] exhibited C–I···N directed infinite one-dimensional chains of alternate ArB and D units. In contrast, C–I···N interactions guide the formation of termolecular complexes in the cocrystal of 4-(phenanthren-9-yl)-2,2′-bipyridine with D (PhenB·D0.5). Successful implementation of C–I···N interactions aided by 2,2′-bipyridine and D enabled the tuning of three-dimensional close packing in planar polyaromatic hydrocarbons into a columnar/lamellar arrangement suitable for optoelectronic devices.
Pyrene-based materials possessing one-dimensional cofacial π–π stacked structural motifs with short interplanar distances can have favorable charge transport properties. In this work, we have realized the switching of sandwich herringbone to lamellar/columnar arrangement by introducing α-haloacetyl substituents in pyrene. Density functional theory calculations showed that the subtle difference in the charge distribution induced by the haloacetyl group plays an important role in regulating the cofacial stacking. Single-crystal X-ray structural analyses and quantum theory of atoms in molecules (QTAIM) analyses revealed the role of dihydrogen contacts, halogen interactions, and hydrogen bonding in extending the columnar/lamellar arrangement in three dimensions. Hirshfeld surface analyses showed γ (lamellar) and β (columnar) packing motifs indicating extensive π–π stacking interaction between pyrene units. Detailed structural analysis revealed a gradual decrease in the intermolecular π–π stacking distance (dπ–π = 3.39 Å) with increasing the polarizability of halogen atom, a characteristic interplanar distance observed in highly oriented pyrolytic graphite (dπ–π = 3.35–3.39 Å).
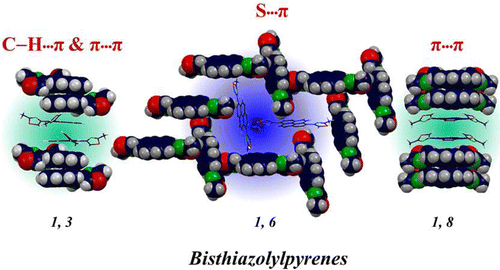
The formation of well-defined packing motifs and control of π–π stacking by rationalizing the effects of molecular substitution in π-conjugated organic materials represent a significant challenge in supramolecular design. To explore the influence of the thiazolyl group in the solid state packing and photophysical properties of pyrene, a new class of three regioisomeric bisthiazolylpyrenes (2-2″TP) were designed and synthesized. Single crystal X-ray structure and quantum theory of atoms in molecules analysis revealed the presence of S···π interactions in 2′TP. Hirshfeld analysis showed the γ and β packing motif in 2TP and 2″TP, respectively, due to extensive π–π interactions, observed S···π interactions led to herringbone packing in 2′TP. The diverse packing arrangement of 2-2″TP led to a notable difference in the fluorescence behavior in the crystalline state. 2′TP possessing S···π interaction showed remarkable enhanced emission in the crystalline state compared to the amorphous state, in contrast to 2TP and 2″TP. Our results provide new insight for the rational design and regulation of intermolecular interactions of conjugated heterocyclic aromatics to achieve a diverse degree of orbital overlap between neighboring chromophore units and thereby favorable optoelectronic properties.
We report the synthesis, X-ray structures and photophysical properties of a few benzoylpyrene (BP) derivatives. Steric hindrance due to incremental benzoyl groups causes a systematic reduction in the orbital overlap (π–π) between vicinal pyrene units affording green-yellow-orange solid-state emitters. Crystallization induced emission could arise from: i) electronic (dipolar/excitonic) interactions, ii) arrested bond rotations, and/or iii) lack of solvation in crystalline 1–4BP (ΦFl ∼ 2–26%) when compared to that in solution (ΦFl ≤ 1%). Our earlier effort [Chem. Commun. 2014, 50, 8644] on progressive acylation, in contrast to benzoylation, results in a gradual increase in the π–π overlap between vicinal pyrenes.
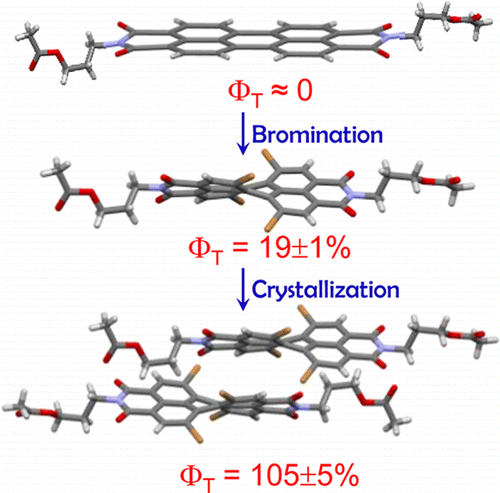
Solvent-free crystal structure of N,N-bis(propylacetyl)-1,6,7,12-tetrabromoperylene-3,4:9,10-bis(dicarboximide), PDI-Br4, obtained by X-ray diffraction reveals the core-twisted perylene motif having π–π stacks at an interplanar separation of 3.7 Å. Slip-stacked arrangement of PDI units in PDI-Br4 arises due to the presence of bulky bromine atoms. Femtosecond pump–probe measurements of monomeric PDI-Br4 in toluene reveal ultrafast intersystem crossing (τISC < 110 fs) when excited at 400 nm. Triplet quantum yield (ΦT) of 19 ± 1% and 105 ± 5% for PDI-Br4 in toluene and vapor-annealed polycrystalline 60 nm thick film respectively are estimated from nanosecond transient absorption measurements. Quantum chemical calculations show that the combined effects of heavy atom and core-twist in PDI-Br4 can activate the intersystem crossing by altering the singlet–triplet energy gap. Enhanced quantum yield accounts for the singlet fission mediated generation of triplet excited state in the PDI-Br4 thin film.
The crystallization of weakly fluorescent 4-amino-2,2′-bipyridine (AMBPY) in solution phase under ambient conditions afforded three fluorescent conformational polymorphs. The marginal increase in the barrier to rotation observed in AMBPY as compared to unsubstituted 2,2′-bipyridine could be attributed to the “buttressing effect” offered by the amino substituent at the meta position. A smaller yet significant difference in energy (0.1–2.6 kJ mol−1) with respect to the global minima facilitates the isolation of AMBPY-I–III polymorphs. A unique nitrogen–nitrogen interaction is observed in two of the polymorphs, namely, AMBPY-I and AMBPY-III, promoted by cooperative C⋯H and N⋯H interactions. A crystallization-induced enhancement (ca. 5–10 fold) in the fluorescence quantum yield of AMBPY polymorphs is observed relative to the solution/amorphous state. Controlling the luminescence properties of molecular solids by tuning their packing arrangements via various interactions is an integral aspect in the construction of novel photo-functional materials.
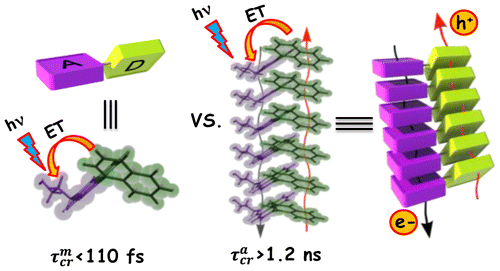
We report a nonparallel stacked arrangement of donor–acceptor (D–A) pairs for prolonging the lifetime of photoinduced charge-separated states. Hydrogen–hydrogen steric repulsion in naphthalimide-naphthalene (NIN) dyad destabilizes the planar geometry between the constituent units in solution/ground state. Sterically imposed nonplanar geometry of the dyad allows the access of nonparallel arrangement of the donor and acceptor stacks having triclinic space group in the crystalline state. Antiparallel trajectory of excitons in nonparallel D–A stacks can result in lower probability of geminate charge recombination, upon photoexcitation, thereby resulting in a long-lived charge-separated state. Upon photoexcitation of the NIN dyad, electron transfer from naphthalene to the singlet excited state of naphthalimide moiety results in radical ion pair intermediates that survive >10,000-fold longer in the aggregated state (τcra > 1.2 ns) as compared to that of monomeric dyad (τcrm < 110 fs), monitored using femtosecond transient absorption spectroscopy.We report a nonparallel stacked arrangement of donor–acceptor (D–A) pairs for prolonging the lifetime of photoinduced charge-separated states. Hydrogen–hydrogen steric repulsion in naphthalimide-naphthalene (NIN) dyad destabilizes the planar geometry between the constituent units in solution/ground state. Sterically imposed nonplanar geometry of the dyad allows the access of nonparallel arrangement of the donor and acceptor stacks having triclinic space group in the crystalline state. Antiparallel trajectory of excitons in nonparallel D–A stacks can result in lower probability of geminate charge recombination, upon photoexcitation, thereby resulting in a long-lived charge-separated state. Upon photoexcitation of the NIN dyad, electron transfer from naphthalene to the singlet excited state of naphthalimide moiety results in radical ion pair intermediates that survive >10,000-fold longer in the aggregated state (τcra > 1.2 ns) as compared to that of monomeric dyad (τcrm < 110 fs), monitored using femtosecond transient absorption spectroscopy.
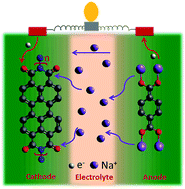
Developing new approaches to improve the performance of organic electrodes for rechargeable sodium batteries is important. Here, we report studies on N,N′-diamino-3,4,9,10-perylenetetracarboxylic polyimide (PI) as a novel cathode for a sodium battery and demonstrate an all-organic sodium ion battery using this polyimide as the cathode and disodium terephthalate (NaTP) (pre-sodiated) as the anode. The synthesised PI exhibits excellent electrochemical properties, when studied as the cathode for sodium batteries, with a reversible capacity of 126 mA h g−1 along with good capacity retention and rate capability, in the voltage range of 1.5 to 3.5 V vs. Na+/Na. The all-organic sodium ion full cell delivered an initial capacity of 73 mA h g−1, with an average cell voltage of 1.35 V. The attractive electrochemical performance combined with the design flexibility of a PTCDA based PI material, offer new possibilities for the development of efficient all-organic sodium ion batteries.
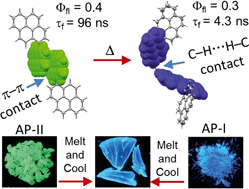
We report the C–HH–C and C–H⋯π interaction assisted formation of thermodynamically stable blue emissive AP-I from the kinetically stable green emissive AP-II crystal polymorph of 1-(anthracen-9-yl)pyrene (AP). The rotational degree of freedom at the bond between the two constituent chromophores facilitates the formation of conformational polymorphs AP-I and AP-II that can be quantitatively crystallized from ethyl acetate and hexane, respectively.
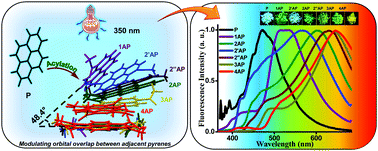
Quantum theory of atoms-in-molecules and Hirshfeld surface analyses indicated an increase in the extent of (i) C–H⋯H–C; (ii) C–H⋯O, (iii) π–π interactions and a decrease in the extent of (i) σ–π interaction, (ii) an interplanar angle between the vicinal pyrene units in a series of acetylpyrene derivatives offering blue–green–orange emissive crystals.
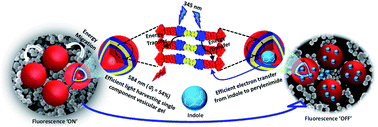
We report the synthesis and excited state properties of a single component light harvesting donor–acceptor dyad (R/S)-NP(OH)2 containing an α,β-dihydroxypropyl side-chain that can undergo molecule–bilayer–vesicle–gel–crystal transition. By virtue of steric hindrance offered by orthogonal naphthalimide and 2,6-diisopropylphenyl substituents, very weak H-type excitonic interactions between the perylenimide units resulted in high fluorescence quantum yield in the dyad-based metastable vesicular gel having a near-quantitative excitation energy transfer from naphthalimide to perylenimide. Femtosecond transient absorption measurements of the dyad (R/S)-NP(OH)2 : indole co-gel show that the vesicular scaffold promoted extension of the survival time of charge separated states (∼1.4 ns) when compared to ultrafast charge recombination (∼6 ps) in dyad (R/S)-NP(OH)2 : indole solution.
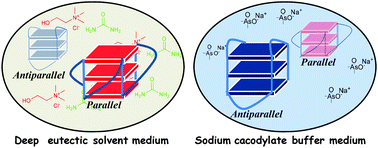
The biocompatibility as well as the sustainability of a deep eutectic solvent makes it a good substitute for aqueous media in studying biomolecules. Understanding the structure and stability of natural and non-natural G-quadruplexes in aqueous and highly viscous media will be useful in biological and nanodevice applications. We report the synthesis and conformational analysis of a model G-rich oligonucleotide G3T3 and non-natural G-rich sequences Pyr1–Pyr3 in aqueous and highly viscous media. Progressive increases in the loop replacement with a non-natural pyrene linker leads to a systematic increase of the thermal denaturation temperature of the modified G-rich oligonucleotides Pyr1–Pyr3 in 10 mM cacodylate buffer (pH 7.2) containing 100 mM KCl, as monitored using UV-Vis spectroscopy. A circular dichroism signal clearly revealed the formation of a predominantly anti-parallel vs. parallel conformation in the natural G-rich oligonucleotide G3T3 as well as the non-natural G-rich oligonucleotides Pyr1–Pyr3 in 10 mM cacodylate buffer (pH 7.2) containing 100 mM KCl. On the other hand, we observed thermodynamic destabilization of G-rich oligonucleotides in a deep eutectic solvent (DES; 1 : 2 choline chloride–urea) containing 100 mm KCl with an increase in loop replacements. Interestingly, we observed an exclusively parallel G-quadruplex conformation in the case of G3T3 in DES containing 100 mm KCl. While pyrene containing G-rich oligonucleotides Pyr1–Pyr3 exhibited a predominantly parallel vs. anti-parallel G-quadruplex conformation in DES containing 100 mM KCl.
The photochemical reactions of eleven synthetic DNA hairpins possessing a single TT step either in a base-paired stem or in a hexanucleotide linker have been investigated. The major reaction products have been identified as the cis–syn (2 + 2) adduct and the (6 − 4) adduct on the basis of their spectroscopic properties including 1D and 2D NMR spectra, UV spectra and stability or instability to photochemical cleavage. Product quantum yields and ratios determined by HPLC analysis allow the behaviour of the eleven hairpins to be placed into three groups: Group I in which the (2 + 2) adduct is the major product, as is usually the case for DNA, Group II in which comparable amounts of (2 + 2) and (6 − 4) adducts are formed, and Group III in which the major product is the (6 − 4) adduct. The latter behaviour is without precedent in natural or synthetic DNA and appears to be related to the highly fluxional structures of the hairpin reactants. Molecular dynamics simulation of ground state conformations provides quantum yields and product ratios calculated using a single parameter model that are in reasonable agreement with most of the experimental results. Factors which may influence the observed product ratios are discussed.
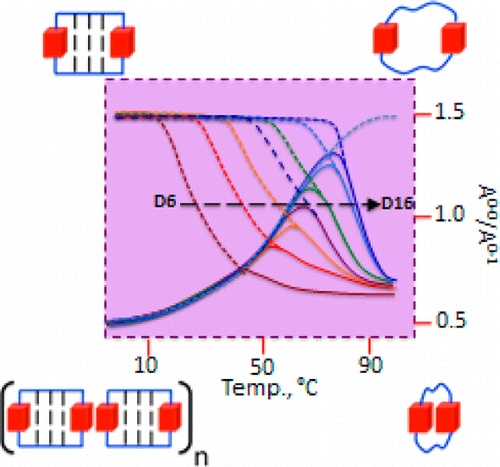
We report the self-assembly and thermal dissociation of DNA dumbbell conjugates having a perylenediimide (PDI) linker on each end separated by 6–16 A–T base pairs. In the presence of NaCl these dumbbells form one-dimensional supramolecular assemblies as a consequence of the hydrophobic association of their PDI sticky ends. The dependence of assembly formation on dumbbell concentration, salt concentration, and temperature can be conveniently monitored by UV–vis spectroscopy. The melting of these linear assemblies follows two limiting mechanisms, depending on the length of the dumbbells. Upon heating in the presence of salt, the assemblies formed by the longer dumbbells undergo a sequential transition from assembly to base-paired monomer to random coiled monomer, whereas the assemblies formed by the shorter dumbbells undergo disassembly and base-pair melting cooperatively. In all cases, the intramolecular hydrophobic association of the PDI chromophores is observed at elevated temperature. The thermal behavior of these one-dimensional assemblies is compared to that of other sticky-ended assemblies.
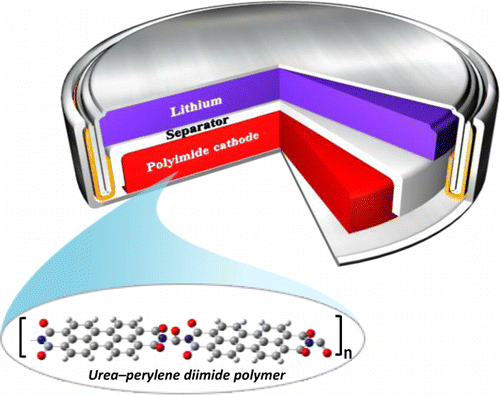
Organic materials for Li-ion battery application continue gaining attention due the virtue of low cost, environmental benignity, and so on. A new class of electroactive organic material called polyimides is particularly important due to the extra stability exhibited at higher current rates. High-performance rechargeable lithium battery cathodes based on polyimides of 3,4,9,10-perylenetetracarboxylicacid-dianhydride are prepared. The novel electrodes exhibit good rate capability and improved cycling stability, which result from their combined beneficial properties such as the presence of additional carbonyl groups, favorable band gap, and enhanced conductivity, making it a potential material for greener and sustainable electrochemical storage devices.
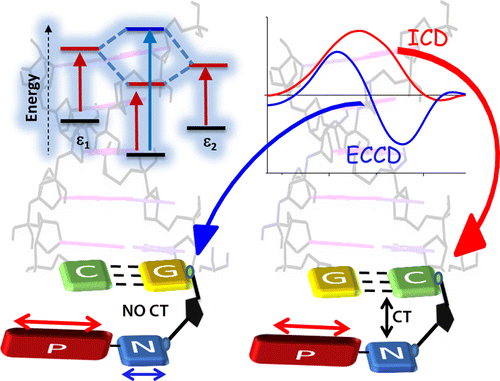
Circular dichroism spectra for a series of structurally analogous hairpin oligonucleotides, tethered at the 5′-end with an axially chiral naphthalenimide-perylenimide dyad (NP), is dependent on the nature (AT vs GC) and the orientation (5′-C vs 5′-G) of the adjacent base pair that stacks with the dyad. Charge transfer (CT) interaction between the naphthalenimide unit of the dyad NP and the adjacent guanine–cytosine (5′-C) base pair has been characterized by UV–vis absorption and fluorescence measurements. Molecular dynamics simulations of the dyad end-capped hairpin DNA ODN6i and TD-DFT calculations of the naphthalenimide and perylenimide units confirm that the CT transition dipole orients perpendicular to the perylenimide transition dipole. The orthogonality of the cross product of the CT and the perylenimide transition dipoles with the displacement vector connecting the two dipoles in space results in the zeroing out of the rotational strength of the perylenimide transition dipole, subsequently leading to the DNA-induced nonexciton coupled circular dichroism corresponding to perylenimide, in concurrence with experimental CD spectrum. Singular value decomposition of thermal denaturation of the hairpin DNA having CT interaction (ODN6i) revealed the denaturation proceeded through an additional intermediary stage compared to the hairpin DNA without the CT interaction (ODN6). Dissimilar CD (induced CD for ODN6i vs exciton-coupled CD for ODN6) spectra corresponding to perylenimide unit obtained for similar NP end-capped hairpin DNA sequences cautions against the indiscriminate use of the exciton chirality method, particularly in systems like DNA and proteins containing polarizable chromophores that can interact with reporter transition dipoles.
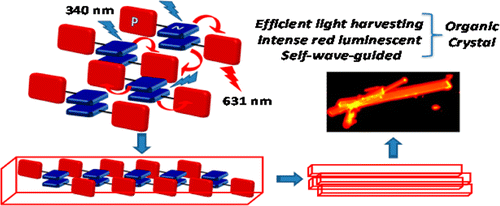
Highly efficient photoinduced energy transfer is observed in an orthogonal bichromophore naphthalenimide-perylenimide (NP), leading to strong solid-state luminescence (Φ = 0.5 ± 0.04) in the red region (λem = 631 nm). Steric hindrance imparted by orthogonal naphthalenimide and diisopropyl phenyl units prevents the association of perylenimide moieties, thereby retaining a high quantum yield of fluorescence emission even in the crystalline state. Upon photoexcitation at 340 nm, the intramolecular parallel orientation of the transition dipoles permits efficient Coulombic coupling between naphthalenimide and perylenimide in combination with weak H-type excitonic interactions between the intermolecular perylenimide units that results in intense red fluorescence in the crystalline state of dyad NP. Negligible photoinduced electron transfer from the singlet excited state of the perylenimide to the naphthalenimide unit (ΔG = 0.44 eV) and a marginal feasibility (ca. 10%) of photoinduced electron transfer from the singlet excited state of the naphthalenimide to the peryleninimide unit (ΔG = −0.17 eV) makes the dyad strongly fluorescent when excited at both 340 and 475 nm, corresponding to the naphthalenimide and perylenimide units, respectively. A narrow emission at 631 nm, wide absorption window (300–600 nm), and high fluorescence quantum yield in the crystalline state make the dyad NP a potential candidate for optoelectronic and photonic applications.
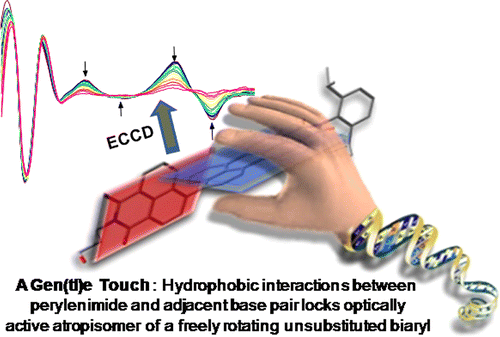
We report the conformational restriction of a freely rotating biaryl derivative resulting in strong exciton coupled circular dichroism between the two nondegenerate fragments of the dyad NP only in the presence of DNA. The napthalenimide–perylenimide dyad NP, an extended 1,1′-binaphthalene derivative, has a rotational barrier of 100 kJ/mol, leading to fast racemization. While end-stacking with a DNA hairpin, NP exhibits a significantly high rotational barrier, an additional 91 kJ/mol, resulting in atroposelective preference. Such reversible conformational constraints using biotemplates show huge potential toward conformational analysis, dynamic kinetic resolution, and asymmetric synthesis. The present work focuses on conformational analysis of a freely rotating biaryl derivative using a combination of techniques such as temperature-dependent UV–vis, circular dichroism, steady-state and time-resolved fluorescence spectroscopy, and molecular dynamics simulation.
With an increase in temperature, an unprecedented restoration of symmetry in the symmetry breaking excited state charge transfer is observed in a geminal pair of near-orthogonally connected perylenimide dimers. Such restoration of symmetry could be attributed to the interchromophoric planarization and/or loss of solvation asymmetry at elevated temperature resulting in enhanced fluorescence quantum yield.
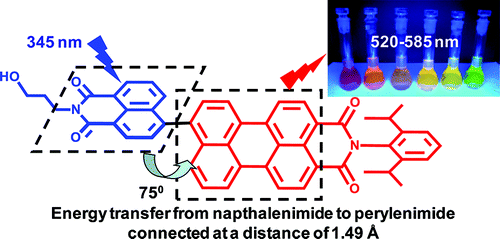
A combined experimental and theoretical study shows a significant barrier (ca. 100 kJ/mol) to rotation through the interchromophoric carbon–carbon single covalent (1.49 Å) bond between the naphthalenimide and perylenimide units that prevents coplanarization of the two units in the dyad NP, thereby forcing them to act as independent chromophores/redox centers. Upon photoexcitation, highly efficient energy transfer is observed from the naphthalenimide (energy donor) to the perylenimide (energy acceptor) moiety predominantly through Coulombic coupling, completely isolating the orbital overlap (Dexter-type) interaction between the chromophoric units at such short separation by virtue of their orthogonal arrangement. Because Förster’s ideal-dipole approximation ignores the contribution from significant higher-order Coulombic interactions at such short distances between donor and acceptor moieties, the complete coupling was computed from the transition densities, giving an estimate of the energy-transfer rate from the naphthalenimide donor to the perylenimide acceptor of kET = 2.2 × 1010 s–1, in agreement with observations. Ultrafast excitation energy (ca. 40 ps, 90%) and electron (<0.5 ps, 10%) transfer from the singlet excited state of naphthalenimide to the perylenimide moiety competes with further delayed processes in the conjugate NP. Upon excitation at 345 nm, conjugate NP exhibits near-quantitative energy transfer in conjunction with solvent-polarity-dependent (solvatochromic) perylenimide fluorescence, resulting in a remarkable Stoke’s shift of ca. 175–240 nm. Favorable photophysical properties such as high fluorescence quantum yield, wide excitation range, ultrafast energy transfer, marginal electron transfer, and large Stoke’s shift make this conjugate a potential candidate for biological applications.
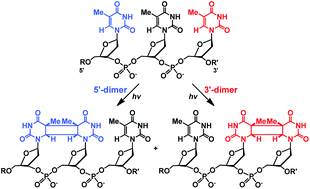
Irradiation of alkane-linked DNA hairpins possessing TTT steps with flanking purine bases yields products identified as the cis–syn (2 + 2) dimers formed between the central thymine and its 3′- and 5′-neighbors. Selective formation of the 3′-dimer is attributed to ground state conformational effects and electron transfer quenching by purine bases.
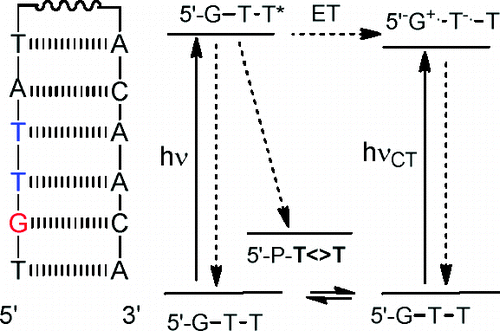
Quantum yields for thymine photodimerization (ΦTT) have been determined for a series of short DNA single-strand and base-paired hairpin structures possessing a single thymine–thymine step with flanking purines. Values of ΦTT are strongly dependent upon the oxidation potential of the flanking purine, decreasing in the order: inosine > adenine > guanine > deazaguanine. The dependence of ΦTT on the ionization potential of the flanking purine is more pronounced when the purine of lower oxidation potential is located at the 5′- versus 3′-position in either a single strand or a hairpin. Molecular dynamics simulations for hairpin structures indicate that the TT step is π-stacked with both the 5′ and 3′ purine, but that there is little π-stacking with either purine in single-strand structures. The observation of moderately intense long-wavelength UV absorption features for hairpins having 5′-Z or G flanking purines suggests that excitation of ground state donor–acceptor complexes may account for more extensive quenching of dimerization by 5′- versus 3′-purines. The “purine effect” on ΦTT is attributed to a combination of ground state conformation, ground state electron donor–acceptor interactions, and excited state exciplex formation.
Alkane linkers derived from α,ω-alkane diols have been employed for the preparation of DNA mini-hairpins, which are suitable for studies of the spectroscopy and photochemistry of short base pair domains. The solution structure of a DNA hairpin having a n-dodecane linker and six AT base pairs has been determined using 1H NMR data with restrained molecular dynamics. The chemical shifts of the 12 diastereotopic pairs of linker protons show a distribution of values depending on their locations within the shielding region of the ring-currents of the adjacent base pair. Chemical shifts calculated from the mean NMR solution structure show the same overall distribution, but with larger absolute values for the upfield shifts. The C12 linker lies approximately parallel to the adjacent base pair and adopts a highly curved structure having an average of 4−5 gauche C−C bonds, in contrast to a modeled B-DNA structure which has a more fully extended alkane chain. This curved structure results in an average P−P distance of 15.4 Å, substantially shorter than the 17.7 Å average distance for B-DNA. This structure appears to maximize the hydrophobic interactions between the linker and the adjacent base pair. Partial NMR assignments for the C12 linker in a second hairpin having a different base-pair sequence indicates that it adopts a similar curved geometry having multiple gauche bonds. The hairpin base pair domain adopts a B-DNA geometry in which the AT base pair adjacent to the linker displays Watson−Crick base pairing, in spite of the relatively short P−P distance. The terminal bases-pair shows evidence of extensive end-fraying. The four interior base pairs display a normal B-DNA geometry, thus providing good models for base pair domains embedded in longer duplexes.
The self-assembly of DNA dumbbell conjugates possessing hydrophobic perylenediimide (PDI) linkers separated by an eight-base pair A-tract has been investigated. Cryo-TEM images obtained from dilute solutions of the dumbbell in aqueous buffer containing 100 mM NaCl show the presence of structures corresponding to linear end-to-end assemblies of 10−30 dumbbell monomers. The formation of assemblies of this size is consistent with analysis of the UV−vis and fluorescence spectra of these solutions for the content of PDI monomer and dimer chromophores. Assembly size is dependent upon the concentration of dumbbell and salt as well as the temperature. Kinetic analysis of the assembly process by means of salt-jump stopped-flow measurements shows that it occurs by a salt-triggered isodesmic mechanism in which the rate constants for association and dissociation in 100 mM NaCl are 3.2 × 107 M−1s−1 and 1.0 s−1, respectively, faster than the typical rate constants for DNA hybridization. TEM and AFM images of samples deposited from solutions having higher concentrations of dumbbell and NaCl display branched assemblies with linear regions >1 μm in length and diameters indicative of the formation of small bundles of dumbbell end-to-end assemblies. These observations provide the first example of the use of hydrophobic association for the assembly of small DNA duplex conjugates into supramolecular polymers and larger branched aggregates.
The results of an integrated experimental and theoretical study of thymine−thymine photodimerization in short single-strand and duplex DNA structures possessing a single locked nucleic acid TT step are reported. Control of ground-state conformation by the locked nucleic acids results in a marked increase in both the quantum yield and the selectivity of photoproduct formation.
The synthesis and properties of three related families of alkane-linked DNA hairpins are reported. The first possesses a dodecane linker (C12) and 2−8 AT base pairs. The second possesses six AT base pairs and straight chain alkane linkers having 8−16 methylenes. The third has three alkane linkers of different length and a constant six base-pair stem with alternating A-T bases and a single TT step. The spectroscopic properties (UV, CD, and 1H NMR) and molecular modeling are consistent with the formation of base-paired B-DNA structures for all hairpins having four or more AT base pairs. The thermal stability of hairpins having a C12 linker is greater than that of the commonly used hexa(ethylene glycol) linker but less than that of the stilbenediether linker having the same AT base-pair domain. Hairpin stability is related to both hydrophobic interactions between the linker and the adjacent base pair (stilbene > alkane > glycol) and the overall length of the linker. The stability of the alkane-linked hairpins having six AT base pairs is greater for a tetradecane linker than for either shorter or longer linkers. The good thermal stability of alkane-linked hairpins and absence of a chromophore which absorbs in the UV region makes them well-suited for studies of the electronic spectra and photochemistry of short hairpins having variable base-pair sequences.
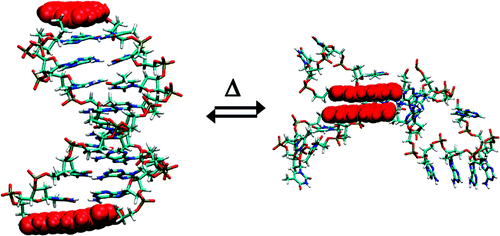
The synthesis and properties of synthetic DNA dumbbells having A-tract base pair domains consisting of 6−16 base pairs connected by perylenediimide (PDI) linkers are reported. The dumbbells were prepared in good yield by chemical ligation of nicked-dumbbell precursors having six or more A-T base pairs. The dumbbell structures have been investigated by a combination of electronic spectroscopy and molecular dynamics simulations. UV−visible spectra are indicative of the formation of monomeric dumbbells at room temperature in aqueous buffer in the absence of added salt. The long-wavelength region of the circular dichroism (CD) spectra is a composite of induced CD of the PDI monomers and intramolecular exciton-coupled CD between the two PDI chromophores. Weak exciton coupled CD can be observed between PDI chromophores separated by 13 base pairs, thus extending the reach of dumbbell molecular rulers. Upon heating, the dumbbells undergo base pair melting and intramolecular PDI−PDI association. Analysis of the temperature-dependent spectral data provides evidence for a three-state model in which base pair melting of the intact dumbbell results in the formation of an intermediate species which is in equilibrium with a collapsed dumbbell having intramolecular PDI−PDI stacking. The low melting temperatures of the shorter dumbbells are attributed to the partial compensation of PDI−PDI association for base pair dissociation.
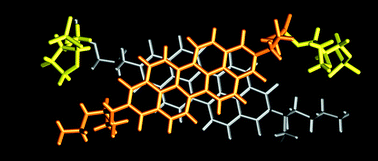
The synthesis, structure, and electronic spectra of a series of DNA hairpins possessing two perylenediimide (PDI) base pair surrogates are reported. The PDI chromophores are located in opposite strands of the hairpin base pair domain opposite abasic sites and are either adjacent to each other or separated by a variable number of AT or GC base pairs. Molecular modeling of the conjugate having adjacent PDI chromophores shows that they adopt a slipped, π-stacked geometry with an angle of 40° between the PDI long axes. The electronic absorption, fluorescence, and circular dichroism of this conjugate are consistent with a stacked PDI structure. Conjugates having one or two GC base pairs between the PDI chromophores display spectra that are consistent with isolated PDIs. Conjugates having 1–4 AT base pairs have more complex spectra, suggestive of an equilibrium between base paired and flipped structures having stacked PDIs. Heating of the conjugates possessing isolated PDI chromophores results in base pair flipping. The free energy for PDI stacking is greater than that for a single AT base pair and comparable to that for a single GC base pair or two AT base pairs.
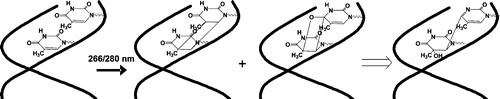
The paper presents quantum yield results for the [2+2] and 6-4 photodimerization of TT steps in several DNA structures, including hairpins where the context dependence of the photodimerization yield is determined, and it develops a theoretical model that correctly describes the trends in dimerization yield with DNA structure. The DNA conjugates considered include dT20, dA20dT20, and three alkane-linked hairpins that contain a single TT step. The theoretical modeling of the [2+2] process is based on CASSCF electronic structure calculations for ethylene + ethylene, which show that photoexcitation of low-lying excited states leads to potential surfaces that correlate without significant barriers to a conical intersection with the ground state surface at geometries close to the dimer structure. The primary constraint on dimerization is the distance d between the two double bonds, and it is found that d < 3.52 Å leads to quantum yield trends that match the observed trends within a factor of 3. Constraints on the dihedral angle between the two double bonds are not as important, and although it is possible to generate better dimerization yield predictions for some structures by including these constraints, the best overall picture is obtained with no constraint. For 6-4 dimerization, a distance g < 2.87 Å and no constraint on dihedral angle provide an accurate description of the yield.
We report the effect of steric factors of a few squaraine dyes, bis(2,4,6-trihydroxyphenyl)squaraine (1), bis(3,5-dibromo-2,4,6-trihydroxyphenyl)squaraine (2), and bis(3,5-diiodo-2,4,6-trihydroxyphenyl) squaraine (3), on their binding with human (HSA) and bovine (BSA) serum albumins employing photophysical, chiroptical, biophysical, and microscopic techniques. These dyes interact with serum albumins very efficiently and exhibit site selectivity, involving synergistic effects of hydrophobic, hydrogen bonding, and electrostatic interactions. The association constants of these complexes have been determined and are found to be 4.9 × 106 and 4.1 × 105 M−1, respectively, for the dyes 2 and 3 with BSA, while HSA showed relatively higher association constants of 6.0 × 106 and 9.9 × 105 M−1. Highly clear distinction in site-selective binding can be ascertained from time-resolved fluorescence, displacement cum fluorimetry, and circular dichroism (CD) studies. The increased affinity toward the major binding site (site II, domain III) over the relatively smaller binding site (site I, domain II) in the serum albumin with the increasing size of the heavy atoms present in 2 and 3 as compared to 1 indicates the importance of steric factors thereby confirming that the dye structure has a predominant role in deciding site selectivity. The distance between the energy donor and acceptor was calculated using Förster theory, which agrees well with the reported site 1 binding agent dansylamine. In contrast, no energy transfer was observed between tryptophan (Trp-214) present in domain II of the albumins and the dyes 2 and 3, indicating that these derivatives bind less efficiently at site I due to steric conatraints but preferentially bind at site II. Laser flash photolysis studies of the dyes 2 and 3 in the presence of HSA exhibited ca. 2.5-fold enhancements in the triplet lifetimes and quantum yields when compared to that obtained in buffer. The uniqueness of these dyes is that they show substituent size-dependent selectivity at site II of serum albumins and signal the event through “turn on” fluorescence intensity as well as enhanced triplet excited state lifetimes and quantum yields, thereby indicating their potential use as NIR noncovalent protein labeling and photodynamic therapeutic agents.
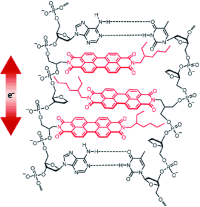
Charge on through: Perylenediimide chromophores incorporated into DNA hairpins serve as base-pair surrogates and form a zipperlike intercalated structure (see picture). Electron hopping was observed within the chromophore stacks upon one-electron reduction.
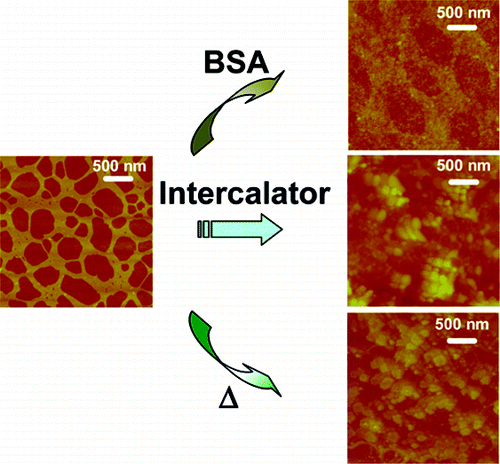
Calf thymus DNA exhibited a regular network-like structure on mica and copper surfaces, respectively, under atomic force (AFM) and scanning electron (SEM) microscopic techniques while oily streak cholesteric birefringent texture was observed on the glass surface under optical polarizing microscopy (OPM). In the presence of an external stimuli such as temperature, intercalating compounds such as the viologen-linked pyrene 1 and para-tolylacridinium iodide (2) and the minor groove binding spermine (4) prevented the DNA−DNA interactions and thereby perturbed the self-assembly of DNA. In contrast, the major groove binding bovine serum albumin (BSA) and the noninteracting ligand ortho-tolylacridinium iodide (3) did not affect the overall morphology of DNA, as characterized through the AFM, SEM, OPM, and circular dichroism (CD) techniques. As far as we know, this is the first report that presents direct evidence for the perturbation of supramolecular assembly of DNA under various conditions and that can be visualized through different microscopic techniques.
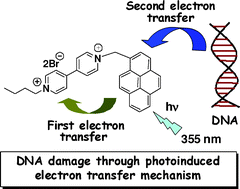
Novel viologen linked pyrene conjugates permeate cells efficiently and exhibit spacer length dependent DNA damage and cytotoxicity upon photoexcitation.
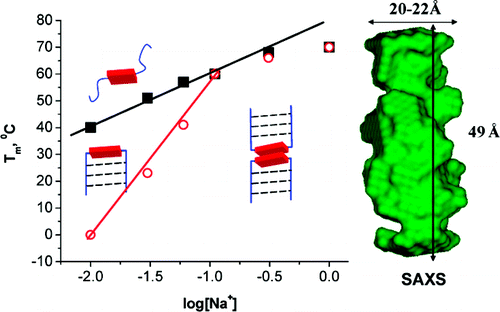
The structure and properties of hairpin-forming bis(oligonucleotide) conjugates possessing perylenediimide (PDI) chromophores as hairpin linkers have been investigated using a combination of spectroscopic and computational methods. These conjugates exist predominantly as monomer hairpins at room temperature in the absence of added salt and as head-to-head hairpin dimers in the presence of >50 mM NaCl. The hairpin dimer structure is consistent with the results of small-angle X-ray scattering in aqueous solution and molecular dynamics simulation. The structure of the nonconjugated PDI dimer in water is investigated using potential of mean force calculations. The salt dependence is attributed to increased cation condensation in the hairpin dimer vs monomer. Upon heating at low salt concentrations, the hairpin dimer undergoes sequential dissociation to form the monomer hairpin followed by conversion to a random coil structure; whereas at high salt concentrations both dissociation processes occur over the same temperature range. The monomer and dimer hairpins have distinct spectroscopic properties both in the ground state and excited singlet state. The UV and CD spectra provide evidence for electronic interaction between PDI and the adjacent base pair. Low fluorescence quantum yields are observed for both the monomer and dimer. The transient absorption spectrum of the dimer undergoes time-dependent spectral changes attributed to a change in the PDI−PDI torsional angle from ca. 20° in the Franck−Condon singlet state to ca. 0° in the relaxed singlet state, a process which occurs within ca. 40 ps.
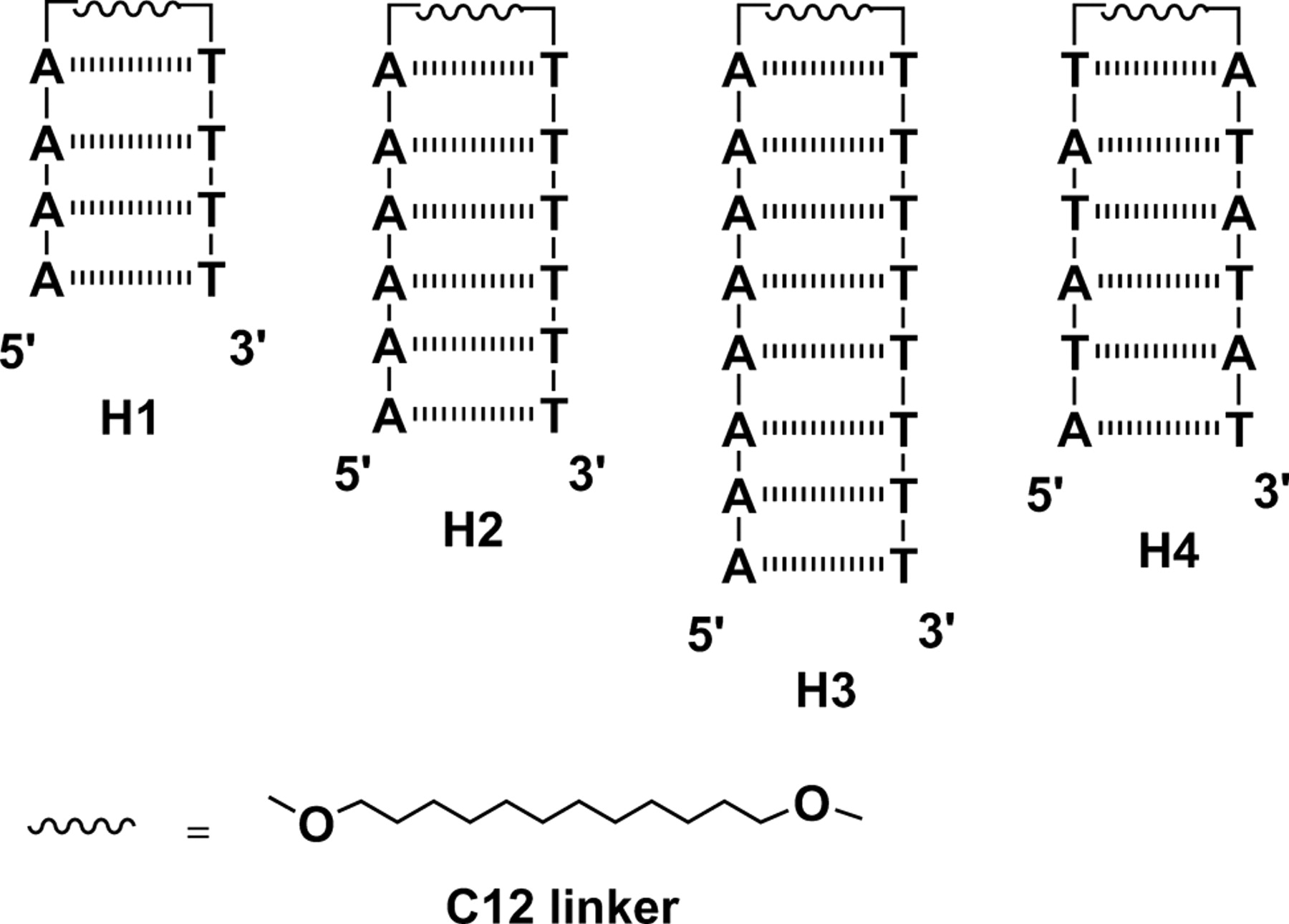
Optical spectra of biological polymers contain important information about their structure and function in living organisms. This information can be accessed by extracting an optical interaction of monomers, i.e., their exciton coupling, from experimental data. This coupling is sensitive to molecular structure, geometry, and conformation and can be used to characterize them. However, the accurate determination of exciton coupling in important biological molecules is difficult because inhomogeneous broadening smears out the monomer interaction. We suggest a way to overcome this problem by applying exact sum rules. These sum rules are derived by establishing a straightforward relationship between integral characteristics of absorption and circular dicroism spectra, and exciton coupling. Exciton coupling between AT pairs in native DNA conformation is estimated by applying these sum rules to DNA hairpin optical spectra as V0 ∼ 0.035 eV in agreement with the earlier numerical calculations.
The structure and properties of hairpin-forming bis(oligonucleotide) conjugates possessing perylenediimide (PDI) chromophores as hairpin linkers have been investigated using a combination of spectroscopic and computational methods. These conjugates exist predominantly as monomer hairpins at room temperature in the absence of added salt and as head-to-head hairpin dimers in the presence of >50 mM NaCl. The hairpin dimer structure is consistent with the results of small-angle X-ray scattering in aqueous solution and molecular dynamics simulation. The structure of the nonconjugated PDI dimer in water is investigated using potential of mean force calculations. The salt dependence is attributed to increased cation condensation in the hairpin dimer vs monomer. Upon heating at low salt concentrations, the hairpin dimer undergoes sequential dissociation to form the monomer hairpin followed by conversion to a random coil structure; whereas at high salt concentrations both dissociation processes occur over the same temperature range. The monomer and dimer hairpins have distinct spectroscopic properties both in the ground state and excited singlet state. The UV and CD spectra provide evidence for electronic interaction between PDI and the adjacent base pair. Low fluorescence quantum yields are observed for both the monomer and dimer. The transient absorption spectrum of the dimer undergoes time-dependent spectral changes attributed to a change in the PDI−PDI torsional angle from ca. 20° in the Franck−Condon singlet state to ca. 0° in the relaxed singlet state, a process which occurs within ca. 40 ps.
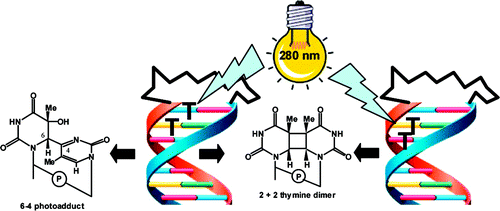
The effect of 280 nm irradiation on a family of synthetic DNA hairpins possessing an alkane linker connecting a six-base pair stem having a single T−T step located at different positions within the hairpin has been investigated. A single adduct assigned to the product of 2+2 dimerization is obtained except in the case of a T−T step located adjacent to the linker, in which case both 2+2 and 6−4 adducts are obtained. The efficiency of dimerization is similar for three hairpins having a T−T step located within the duplex interior. Lower efficiency is observed for a T−T step located at the open end of the hairpin and in T overhangs, whereas higher efficiency is observed for the T−T step adjacent to the linker and in a single T bulge. The context-dependence of dimerization efficiency is discussed.
Interaction of β-cyclodextrin (β-CD) with a few novel electron donor acceptor dyads 1a−c and 2a−c, having aryl and flexible methylene spacer groups, has been investigated through photophysical, chiroptical, electrochemical, NMR, and microscopic techniques. Dyads 1a and 1c, with p-tolyl and biphenyl spacer groups, respectively, exhibited significantly decreased fluorescence quantum yields and lifetimes in the presence of β-CD, while negligible changes were observed for dyad 1b with an o-tolyl spacer. In contrast, spacer-length-dependent significant enhancement in fluorescence quantum yields and lifetimes was observed for dyads 2a−c, with flexible polymethylene (n = 1, 3, 11) spacer groups. Association constants of β-CD encapsulated complexes have been determined and the contrast behavior observed in these systems is explained through an electron transfer (kET) mechanism based on calculated favorable change in free energy (ΔGET = −1.27 eV) and the redox species characterized through laser flash photolysis studies. Rates of kET have been estimated and are found to increase ca. 2-fold in the case of dyads 1a and 1c when encapsulated in β-CD, while significantly decreased kET values were observed for the dyads 2a−c with flexible spacer (ca. 9-fold for 2c). As characterized through cyclic voltammetry, 2D NMR [correlated (COSY) and nuclear Overhauser enhancement (NOESY) spectroscopy], and laser flash photolysis studies, the β-CD encapsulation of dyads with aliphatic spacer groups leads to the conformational unfolding of a sandwich type of structure, whereas dyads with rigid aryl spacer groups undergo unusual planarization as compared to the uncomplexed dyads, resulting in enhanced electron-transfer reaction between the donor and acceptor moieties.
We prepared novel cholesterol-appended squaraine dye 1 and model squaraine dye 2 and investigated their aggregation behavior in solution and thin films using photophysical, chiroptical, and microscopic techniques. Investigations on the dependence of aggregation on solvent composition (good/poor, CHCl3/CH3CN) demonstrated that squaraine dye 1 forms two novel H-type chiral supramolecular assemblies with opposite chirality at different good/poor solvent compositions. Model compound 2 formed J-type achiral assemblies under similar conditions. The supramolecular assembly of 1 observed at lower fractions of the poor solvent could be assigned to the thermodynamically stable form, while a kinetically controlled assembly is formed at higher fractions of the poor solvent. This assignment is evidenced by temperature- and concentration-dependent experiments. With increasing temperature, the chirality of the kinetically controlled aggregate was lost and, on cooling, the aggregate with the opposite chirality was formed. On further heating and cooling the aggregates thus formed resulted in no significant changes in chirality, that is they are thermodynamically stable. Similarly, at lower concentrations, the thermodynamically stable form exists, but at higher concentration aggregation was found to proceed with kinetic control. Based on these observations it can be assumed that formation of the kinetically controlled assembly might be largely dependent on the presence of the nonpolar cholesterol moiety as well as the amount of poor solvent present. However, under solvent-free conditions, structurally different aggregates were observed when drop cast from solutions containing monomer, whereas a left-handed CD signal corresponding to the thermodynamically controlled assemblies was observed from pre-aggregated solutions.
A novel donor−acceptor conjugate 1 was synthesized, and its interactions with various amino acids have been investigated as compared to the model system 2. The conjugate 1 unusually forms an intramolecular charge-transfer complex in the aqueous medium and undergoes selective binding interactions with tryptophan. The uniqueness of this system is that it selectively recognizes tryptophan among all other amino acids and involves synergistic effects of π-stacking, electrostatic, and donor−acceptor interactions.
With the objective of developing efficient DNA oxidizing agents, a new series of viologen-linked pyrene conjugates with the general formula PYLnV2+, having a different number of methylene spacer units (Ln) was synthesized, and their interactions with nucleosides and DNA have been investigated through photophysical and biophysical techniques. The viologen-linked pyrene derivatives PYL1V2+ (n = 1), PYL7V2+ (n = 7), and PYL12V2+ (n = 12) exhibited characteristic fluorescence emission of the pyrene chromophore centered around 380 nm but with significantly reduced yields when compared to those of the model compound PYL1Et3+. The fluorescence quenching observed in these systems is explained through an electron-transfer mechanism based on a calculated favorable change in free energy (ΔGET = −1.59 eV), and the redox species characterized through laser flash photolysis studies. Intramolecular electron-transfer rate constants (kET) were calculated from the observed fluorescence yields, and the singlet lifetimes of the model compound and are found to decrease with increasing spacer length. The DNA binding studies of these systems through photophysical, chiroptical, and viscometric techniques demonstrated that these systems effectively undergo DNA intercalation with association constants (KDNA) in the range of 1.1−2.6 × 104 M-1 and exhibit 2:1 sequence selectivity for poly(dG)·poly(dC) over poly(dA)·poly(dT). Photoactivation of these systems initiates electron transfer from the singlet excited state of the pyrene chromophore to the viologen moiety followed by an electron transfer from DNA to the oxidized pyrene. This results in the formation of stable charge-separated species such as radical cations of both DNA and reduced viologen as characterized by laser flash photolysis studies and subsequently the oxidized DNA modifications. These novel systems are soluble in buffer media, stable under irradiation conditions, and oxidize DNA efficiently and selectively through a cosensitization mechanism and hence can be useful as photoactivated DNA cleaving agents.
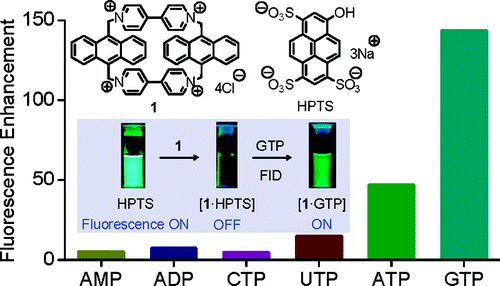
With the objective of developing small molecule based receptors for nucleosides and nucleotides, interactions of a cyclic donor−acceptor conjugate 1 with adenosine, AMP, ADP, CTP, UTP, ITP, ATP, and GTP have been investigated by absorption, steady-state, and time-resolved fluorescence, cyclic voltammetry (CV), NMR, and fluorescence indicator displacement techniques. Titration of 1 with the fluorescent indicator, 8-hydroxy-1,3,6-pyrene trisulfonate (HPTS), resulted in nearly complete fluorescence quenching of HPTS, along with 25% hypochromicity in its absorption spectrum. Benesi−Hildebrand analysis gave a 1:1 stoichiometry for the complex between the receptor 1 and HPTS with an association constant (Kass) of 4.66 × 104 M-1 in buffer. The driving force for such a complexation was evaluated to be the synergistic effects of π-stacking and electrostatic interactions inside the cavity as confirmed by the effect of ionic strength, temperature, and the negative results obtained with the model compound 2. Titration of the nonfluorescent complex [1·HPTS] with various nucleosides and nucleotides resulted in revival of fluorescence of the indicator, HPTS. It was observed that GTP induces maximum displacement of HPTS from the complex [1·HPTS] with an overall fluorescence enhancement of ca. 150-fold. The addition of adenosine, AMP, ADP, CTP, and UTP showed negligible changes, whereas ca. 45- and 50-fold enhancement was observed with ATP and ITP, respectively. The competitive displacement of the indicator by various analytes is found to be in the order GTP (buffer) ≈ GTP (biofluid) ≫ ITP ≈ ATP > UTP > CTP ≈ ADP ≈ AMP ≈ Ade. By virtue of having a better π-electron cloud, GTP undergoes effective electronic, π-stacking, and electrostatic interactions inside the cavity and forms a stable complex with the receptor 1. The uniqueness of this assay is that it differentiates GTP from ATP and other nucleotides and signals the event through a visual “turn on” fluorescence mechanism in buffer as well as in biological fluids.
With the objective of developing small molecule based probes for proteins, interactions of polyhydroxyl-substituted squaraine dye (SQ) with bovine serum albumin (BSA) have been investigated by absorption, steady-state and time-resolved fluorescence, circular dichroism (CD), cyclic voltammetry (CV), 1H NMR, scanning electron, and tapping mode atomic force microscopic techniques. Increase in addition of BSA resulted in increase in absorbance and fluorescence quantum yields (80-fold) of SQ, along with significant bathochromic shifts in the absorption and fluorescence maxima. Half-reciprocal analysis of the absorption data gave a 1:1 stoichiometry for the complex between BSA and SQ with high association (Kass) constant of (1.4 ± 0.1) × 106 M-1 and change in free energy of −35 kJ/mol. The complex formation was further confirmed by observation of induced CD signal corresponding to the SQ chromophore at 610 nm, upfield shift (about Δδ 0.1 ppm) of aromatic protons of SQ in 1H NMR spectra, and decrease in current intensity (CV) of SQ when bound to BSA. The picosecond time-resolved fluorescence studies indicated that the BSA−SQ complex exhibits biexponential decay with significantly enhanced lifetimes of 0.5 and 1.5 ns when compared to the lifetime of SQ (τ = 121 ps) in the absence of BSA. Employing displacement cum fluorimetry using site-specific binding ligands, such as dansylproline and dansylamide, indicated that SQ binds with protein selectively at site II involving hydrophobic, hydrogen bonding, and electrostatic interactions. The uniqueness of this molecular system is that it interacts with BSA selectively at site II and signals the binding event through dual mode recognition of “visual color” change and “turn on” fluorescence mechanism.
Novel cyclophane 1 was synthesized, and its interactions with phosphate, adenosine, AMP, ADP, and ATP have been investigated. With addition of ATP, significant decrease in absorbance of 1 was observed, whereas other guest molecules showed negligible effect. The complex between 1 and ATP was confirmed through cyclic voltammetry and 1H NMR. The uniqueness of the system is that it complexes selectively with ATP in a cavity and involves synergistic effects of both electrostatic and π−π stacking interactions.




























Department of Science and Technology Nano Mission
Dipolar and Multipolar Interactions in Assembled Molecules and Nanostructures: Developing a General Description and its Applications, 01/06/2016-31/05/2019, Rs. 5,61,20,800. Collaborative grant for Prof. K. George Thomas, Dr. R. S. Swathi, Dr. Adithya Lakshmanna and myself.

Indo-Italian Executive Programme of Cooperation in Scientific and Technological Cooperation
Charge and Energy Transfer in Molecular Multifunctional Materials, 16/11/2017-15/11/2019, Rs. 10,20,000/-. Indo-Italian Joint Project for Dr. Mahesh Hariharan, Prof. K. George Thomas and Dr. R. S. Swathi in collaboration with Prof. Anna Painelli from the University of Parma.

Kerala State Council for Science Technology and Environment
Design, synthesis and photocatalytic water splitting properties of functional cobalt based inorganic-organic hybrids, 26/10/2015-15/10/2018, Rs. 45,20,000.

Department of Biotechnology
Mechanistic Investigations on Light Induced Crosslinking of DNA Protein Nanostructures, 15/02/2013-14/02/2016, Rs. 53,76,000
Department of Science and Technology
Synthesis, Structure and Electronic Properties of Natural and Non-Natural Nucleic Acid Sequences, 24/05/2012-13/05/2015, Rs. 26,08,000

Royal Society of Chemistry, UK
Awarded a travel grant of Rs. 1,00,000 (£ 1000) to attend ‘2nd International Conference on Clean Energy Science’ Qingdao, China, April 13-16, 2014.
Kerala State Young Science Scheme
Awarded a travel grant of Rs. 1,70,000 to attend ‘24th Winter I-APS Conference’ Florida, USA, January 1-4, 2015

Indo-US Science and Technology Forum
Awarded a fellowship/travel grant of Rs. 6,26,000 to visit Montana State University, USA, June 9-September 5 2014

Royal Society UK and DST India
Awarded a travel grant of Rs. 1,00,000 to attend ‘Indo-UK Scientific Seminar’, University of Leeds, UK, February 16-18, 2015
You can find Mahesh at his office located at Room No. 3112, Chemical Science Block,IISER Thiruvananthapuram. He is at his office every day from 9:00 am until 7:00 pm, but you may consider a call/email to fix an appointment.
Postdoctoral position is available for highly qualified young scientists to carry out research in our group at the Indian Institute of Science Education and Research Thiruvananthapuram. It is a finite term, contractual, and fulltime position. The initial appointment will be for one year and extendable up to one more year on the basis of performance.
• The Fellowship carries a remuneration of INR 55,000/, with no other allowances.
Eligibility
● Candidates who have a Ph.D. degree in Chemical Sciences are eligible to apply. Those who have submitted the thesis are also eligible to apply. If selected, they have to produce Ph. D. degree certificate or Ph. D. thesis submission certificate from their parent institution.
● The upper age limit is 32 years for general candidates. Relaxation in age will be given for OBC-NCL (34) and SC/ST (35) candidates as per GoI norms.
Application
● The application must be submitted via email to mahesh@iisertvm.ac.in on or before 24th November 2019
● Candidate must submit the following PDF files to mahesh@iisertvm.ac.in
Curriculum Vitae including names of two referees and their email ID & phone no.
Candidate is required to ask at least two referees to submit the confidential letters of recommendation by email to mahesh@iisertvm.ac.in
Research Proposal (maximum two pages)
Thesis summary (maximum two pages)

























































































{kind=link}